Медицинские интернет-конференции
Языки
Электронейромиография и ДНК-диагностика как основа современного медико-генетического консультирования при наследственной моторно-сенсорной невропатии (клиническое наблюдение)
Аксенова А.А., Кузнецова М.А., Павёлкина Е.И., Ситкали И.В.
Резюме
Ключевые слова
Статья
Научный руководитель: д.м.н., доцент Колоколов О.В.
Введение. Болезнь Шарко-Мари-Тута (невральная амиотрофия Шарко-Мари, наследственная моторно-сенсорная невропатия (НМСН)) объединяет группу генетически гетерогенных заболеваний периферической нервной системы (ПНС), характеризующихся симптомами прогрессирующей полинейропатии с преимущественным поражением мышц дистальных отделов конечностей. НМСН является самым частым среди наследственных заболеваний ПНС. Распространенность её в различных популяциях варьирует от 10 до 40 случаев на 100 тысяч населения. В настоящее время известно более 40 локусов и 20 генов, мутации в которых ответственны за развитие фенотипа НМСН.
Диагностика НМСН основывается на клинических данных, результатах электронейромиографии (ЭНМГ), а также ДНК диагностики, позволяющей не только достоверно определить тип мутации, но и провести медико-генетическое консультирование.
Все НМСН по результатам ЭНМГ и морфологическим признакам делят на три основных типа: 1) демиелинизирующий (НМСН I), характеризующийся снижением скорости проведения импульса (СПИ) (СПИ по срединному нерву 38 м/с), 3) промежуточный вариант со СПИ по срединному нерву от 25 до 45 м/с. ЭНМГ исследование имеет важнейшее значение для последующей ДНК-диагностики, так как позволяет определить оптимальный алгоритм молекулярно-генетического обследования для каждого пациента с наименьшими затратами времени и средств.
Несмотря на достижения нейрогенетики в диагностике НМСН, выявление этого заболевания очень часто происходит уже на поздних стадиях, когда эффективность лечебных и реабилитационных мероприятий низкая. Трудности ранней клинической диагностики связаны с малосимптомным течением болезни, фенотипическим полиморфизмом и генетической гетерогенностью. В настоящее момент лечение НМСН носит симптоматический характер.
Наиболее важной задачей лечения является сохранение моторных функций. Для этого рекомендуют индивидуально подобранную физиотерапию и лечебную гимнастику, но учитывают, что больным следует избегать перенапряжения. Для исправления проблем, вызванных деформацией стоп, могут использоваться специальные крепления, которые помогают контролировать тыльное сгибание стопы и голени, нестабильность голеностопного сустава и, зачастую, обеспечивают лучшее чувство равновесия. Аномалии ходьбы могут быть исправлены путем использования разных типов подтяжек (ankle-foot orthoses). Ортопедическая обувь также является важным направлением коррекции походки. Больных должен наблюдать подиатр. По мнению ряда ортопедов, определенный успех имеет хирургическая коррекция деформации стоп.
Клинический пример. Под нашим наблюдением находится пациент А., 33 лет, который предъявлял жалобы на слабость в мышцах дистальных отделов конечностей, деформацию стоп, нарушение походки, затруднения при передвижении в темное время суток. Оказалось, что с детства у него имелась особенность строения стоп (высокий свод), однако никаких затруднений в связи с этим он не испытывал. С 1996г. (16 лет) после перенесенной травмы (вывих правого голеностопного сустава) стала беспокоить слабость и утомляемость в мышцах дистальных отделов ног, что в то время связали с последствиями травмы. Спустя 10 лет (с 2006г.) стали заметными выраженные деформации стоп, нарушилась походка. В 2010г. присоединилась слабость в руках, нарушение чувствительности в стопах, в связи с чем стал затруднятся при передвижении в темное время суток. Однако к неврологам пациент не обращался. В сентябре 2012г. с описанными выше жалобами обратился к ортопедам (ФГБУ «СарНИИТО» МЗ РФ), которые заподозрили НМСН. Диагноз был подтвержден неврологом после проведения ЭНМГ. С целью коррекции грубой деформации стоп в ФГБУ «СарНИИТО» МЗ РФ произведен артродез правого голеностопного сустава.
В настоящее время в неврологическом статусе определяется симметричный дистальный периферический тетрапарез, более выраженный в ногах; нарушение чувствительности по полиневритическому типу; деформация стоп по типу Фридрейха; нарушения координации по типу сенситивной атаксии. Вышеописанное обуславливает выраженные нарушения походки.
При стимуляционной ЭНМГ обнаружены признаки грубого демиелинизирующего поражения моторных и сенсорных волокон периферических нервных стволов конечностей (с резким снижением СПИ), подтверждающее диагноз НИСН.
На основании анализа клинических данных и результатов ЭНМГ диагностирована НМСН Шарко-Мари-Тута I типа.
При обращении пациента проведено медико-генетическое консультирование, в рамках которого обсуждался вопрос о риске рождения у больного А. ребенка, который может заболеть НМСН.
При анализе родословной семьи А. (рис.1) оказалось, что признаки НМСН имеются не только у пробанда, но и у дяди по линии матери. Проявления заболевания у других сибсов, равно как и у других родственников, включая родителей пробанда, отсутствовали, что затруднило определение типа наследования. При аутосомно-доминантном типе наследования вероятность рождения ребенка (любого пола), который заболеет НМСН, составляет 50%. При Х сцепленном доминантном типе наследования все дочери больного отца будут больны, а сыновья – здоровы.
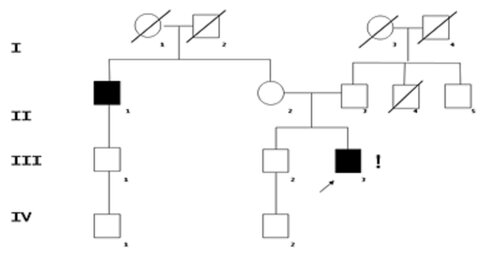
Рис. 1. Родословная
Молекулярно-генетические исследования проведены в лаборатории ДНК-диагностики МГНЦ РАМН. По результатам ДНК-анализа у пациента обнаружена наиболее частая при НМСН I типа мутация – дупликация на хромосоме 17p11.2-p12 (локус ШМТ1А). Таким образом, диагноз моторно-сенсорной нейропатии I типа окончательно подтвержден молекулярно-генетическими методами.
При проведении медико-генетического консультирования установлено, что ребенок, родившийся у пробанда, может заболеть НМСН с вероятностью 50 %.
Заключение. Особенностями данного клинического случая являются поздняя диагностика заболевания; быстро прогрессирующее течение с развитием деформации стоп и ранней инвалидизации, что потребовало проведения хирургической ортопедической коррекции. Кроме того, нетипичным является отсутствие признаков заболевания у родителей пробанда при аутосомно-доминантном типе наследования НМСН.
Выводы. Для обеспечения раннего выявления и профилактики инвалидизации при НМСН необходимо шире использовать методы молекулярно-генетической диагностики; повысить настороженность практикующих врачей в отношении данной патологии; обеспечить междисциплинарных подход в диагностике и ведении пациентов с НМСН, широко используя, как ортопедическую обувь и ортезы, так и (по строгим показаниям) методы хирургической коррекции деформации стоп.
Медицинские интернет-конференции
Языки
Детекция мутации de novo в гене дистрофина и её значение для медико-генетического консультирования при мышечной дистрофии Дюшенна (клиническое наблюдение)
Муравлева Э.А., Стародубова А.В, Пышкина Н.П., Дуйсенова О.С.
Резюме
Ключевые слова
Статья
Детекция мутации denovo в гене дистрофина и её значение для медико-генетического консультирования при мышечной дистрофии Дюшенна
(клиническое наблюдение)
Муравлева Э.А., Стародубова А.В, Пышкина Н.П., Дуйсенова О.С.
Научный руководитель: д.м.н. доц. Колоколов О.В.
ГБОУ ВПО Саратовский ГМУ им. В.И. Разумовского Минздрава РФ
Кафедра неврологии ФПК и ППС им. К.Н. Третьякова
Введение. Мышечная дистрофия Дюшенна (МДД) относится к наиболее часто встречающимся наследственным нервно-мышечным болезням. Распространенность её составляет 2-5 : 100000 населения, популяционная частота – 1 : 3500 новорожденных мальчиков. Эта форма мышечной дистрофии впервые описана Edward Meryon (1852г.) и Guillaume Duchenne (1861г.).
Заболевание характеризуется Х-сцепленным рецессивным типом наследования и тяжелым, прогрессирующим течением. МДД обусловлена мутацией в гене дистрофина, локус которого локализован на Xp21.2. Около 30% случаев обусловлены мутациями de novo, 70% – носительством мутации матерью пробанда. Дистрофин отвечает за соединение цитоскелета каждого мышечного волокна с основной базальной пластинкой (внеклеточного матрикса) через белковый комплекс, который состоит из многих субъединиц. Отсутствие дистрофина приводит к проникновению избыточного кальция в сарколему (клеточную мембрану). Мышечные волокна подвергаются некрозу, происходит замещение мышечной ткани жировой, а также соединительной.
Современная диагностика МДД основана на оценке соответствия проявлений болезни клинико-анамнестическим и лабораторно-инструментальным (креатин-киназа сыворотки крови (ККС), электронейромиография (ЭНМГ), гистохимическое исследование мышечного биоптата) критериям, генеалогическом анализе и данных молекулярно-генетического исследования.
Проведение медико-генетического консультирования в настоящее время во многих семьях позволяет предупредить рождение больного ребенка. Пренатальная ДНК диагностика на ранних сроках беременности в семьях, имеющих ребенка, страдающего МДД, позволит выбрать дальнейшую тактику для родителей и, возможно, досрочно прекратить беременность в случае наличия заболевания у плода.
В ряде случаев клиническая картина наблюдается у женщин – гетерозиготных носительниц мутантного гена в виде увеличения икроножных мышц, умеренно выраженной мышечной слабости, снижения сухожильных и периостальных рефлексов, по данным параклинических исследований повышается уровень ККС. Кроме того, классические клинические проявления МДД могут возникать у женщин с синдромом Шерешевского-Тернера (генотип 45, ХО).
Клинический пример. В нашей клинике наблюдается мальчик К., 7 лет, который предъявляет жалобы на слабость в мышцах рук и ног, утомляемость при длительной ходьбе. Мама ребенка отмечает у него периодические падения, затруднения при подъеме по лестнице, нарушение походки (по типу «утиной»), трудности при вставании из положения сидя, увеличение икроножных мышц в объеме.
Раннее развитие ребенка протекало без особенностей. В возрасте 3-х лет окружающие заметили нарушения двигательных функций в виде появления трудностей при ходьбе по лестнице, при вставании, ребенок не принимал участия в подвижных играх, стал быстро уставать. Затем изменилась походка по типу «утиной». Наросли трудности при вставании из положения сидя или из положения лежа: поэтапное вставание «лесенкой» с активным использованием рук. Постепенно стало заметным увеличение икроножных и некоторых других мышц в объеме.
В неврологическом осмотре ведущим клиническим признаком является симметричный проксимальный периферический тетрапарез, более выраженный в ногах (мышечная сила в проксимальных отделах верхних конечностей – 3-4 балла, в дистальных – 4 балла, в проксимальных отделах нижних конечностей – 2-3 балла, в дистальных – 4 балла). Походка изменена по типу «утиной». Использует вспомогательные («миопатические») приемы, например вставание «лесенкой». Мышечный тонус снижен, контрактур нет. Гипотрофия мышц тазового и плечевого пояса. «Миопатические» черты, например в виде широкого межлопаточного пространства. Имеется псевдогипертрофия икроножных мышц. Сухожильные и периостальные рефлексы – без достоверной разницы сторон; биципитальные – низкие, триципитальные и карпорадиальные – средней живости, коленные и ахилловы – низкие. На основании клинических данных заподозрена МДД.
Помимо пробанда осмотрены его родители и старшая родная сестра. Ни у кого из родственников пробанда клинических проявлений МДД не наблюдалось. Однако у матери замечено некоторое увеличение икроножных мышц в объеме. По данным генеалогического анализа пробанд является единственным заболевшим в семье. При этом нельзя исключить, что мать ребенка и родная сестра пробанда являются гетерозиготными носительницами мутантного гена (рис. 1).
В рамках медико-генетического консультирования семья К. была обследована на предмет наличия/отсутствия делеций и дупликаций в гене дистрофина. Молекулярно-генетический анализ в лаборатории ДНК-диагностики МГНЦ РАМН выявил у пробанда К. делецию 45 экзона, что окончательно подтверждает установленный клинический диагноз МДД. У матери делеция 45 экзона, выявленная у сына, не обнаружена. У сестры в результате анализа делеция 45 экзона, выявленная у брата, не найдена. Следовательно, у исследуемого мутация, скорее всего, имеет происхождение de nоvo, однако также она может явиться результатом герминального мозаицизма у матери. Соответственно, при мутации de novo риск рождения больного ребенка у матери будет определяться популяционной частотой данной мутации (1:3500, ‹‹1%), что значительно меньше, нежели при Х-сцепленном рецессивном типе наследования (50% мальчиков). Поскольку невозможно полностью исключить, что мутация может явиться результатом герминального мозаицизма, при котором наследование по законам Менделя нарушается, рекомендуется проведение пренатальной диагностики при последующей беременности у матери и сестры пробанда.
Заключение. В настоящее время у врача есть широкий арсенал симптоматических средств, используемых в лечении МДД, однако, несмотря на достижения науки, этиологическое лечение МДД до сих пор не разработано, эффективных препаратов для заместительного лечения при МДД не существует. Согласно недавним исследованиям стволовых клеток, существуют перспективные векторы, которые могут заменить поврежденные мышечные ткани. Однако, в настоящее время, возможно лишь симптоматическое лечение, направленное на улучшение качества жизни больного. В этой связи ранняя диагностика МДД играет важнейшую роль для своевременного проведения медико-генетического консультирования и выбора дальнейшей тактики планирования семьи. Для пренатальной ДНК диагностики исследование с помощью биопсии хориона (CVS) можно проводить на 11-14 неделях беременности, амниоцентез можно использовать после 15 недели, забор крови плода возможен примерно на 18 неделе. Если тестирование будет осуществлено на ранних сроках беременности, возможно досрочное прекращение беременности в случае наличия заболевания у плода. В ряде случаев целесообразно проведение преимплантационной ДНК диагностики с последующим экстракорпоральным оплодотворением.
Выводы. Для обеспечения раннего выявления и профилактики МДД необходимо шире использовать методы молекулярно-генетической диагностики; повысить настороженность практикующих врачей в отношении данной патологии. При мутации de novo риск рождения больного ребенка у матери определяется популяционной частотой мутации гена дистрофина. В случаях носительства мутации матерью пробанда требуется пренатальная или перимплантационная ДНК диагностика с целью планирования семьи.
De novo мутации в генах медиаторного комплекса, вызывающие синдромальную интеллектуальную инвалидность: медиаторпатия или транскриптомопатия?
Предметы
Аннотация
Мутации в X-связанном гене MED12 вызывают как минимум три разных, но тесно связанных между собой субъекта с синдромальной интеллектуальной инвалидностью. Недавно был описан новый синдром, вызванный вредными вариантами MED13L, который демонстрирует сходные клинические проявления, включая умственную отсталость, гипотонию и другие врожденные аномалии.
Методы:
Генотипирование 1256 генов, связанных с нервным развитием, было выполнено секвенированием следующего поколения у трех неродственных пациентов и их здоровых родителей. Клинически значимые результаты были подтверждены традиционным секвенированием.
Результаты:
Заключение:
Фенотипические последствия этих мутаций тесно связаны и / или ранее были описаны в одном или другом гене. Кроме того, MED12 и MED13L кодируют двух близкородственных партнеров модуля медиатор-киназы. Следовательно, мы предлагаем концепцию общего клинического спектра MED12 / MED13L, охватывающую синдром Опитца-Каведжиа, синдром Луджана-Фринса, синдром Охдо, синдром гаплоинфективности MED13L и другие.
Главный
Регуляция экспрессии генов у эукариот является чрезвычайно сложным процессом, который контролируется несколькими механизмами на нескольких стадиях (1). Изменения регуляции транскрипции участвуют в патогенезе многих заболеваний человека, и большинство затронутых сигнальных путей в конечном итоге нацелены на основной механизм транскрипции (2).
MED12 (субъединица 12 комплекса медиатора) и MED13L ( подобная субъединице комплекса медиатора 13) кодируют две субъединицы макромолекулярного комплекса, известного как «ко-активаторный комплекс транскрипции медиатора». Медиатор взаимодействует с общими факторами транскрипции и РНК-полимеразой II для регуляции транскрипции. Это существенный коактиватор, действующий в качестве мостиковой молекулы между факторами транскрипции, связанными с вышестоящими регуляторными элементами ДНК, и механизмом транскрипции. Этот комплекс состоит из четырех отдельных модулей (3, 4): головной, средний и хвостовой модули и все вместе образуют основной комплекс медиатора, который напрямую взаимодействует с РНК-полимеразой II и общими и ген-специфическими факторами транскрипции (5, 6). и четвертый модуль «киназы», который обратимо связывается с основным комплексом, который состоит из MED12, MED13, CDK8 и CCNC или их паралогов MED12L, MED13L и CDK19 (7, 8). Предполагалось, что этот модуль в первую очередь участвует в репрессии транскрипции путем блокирования связывания основного комплекса медиатора с РНК-полимеразой II, поскольку медиаторы, содержащие этот модуль, менее активны, чем при его отсутствии (4, 9). Однако есть все больше свидетельств того, что киназный модуль также участвует в активации транскрипции (10).
Здесь мы сообщаем об одном пациенте с мутацией de novo MED12 и двух пациентах с мутациями de novo MED13L, ранее не описанными. Клинические особенности у этих пациентов и у тех, о которых сообщалось ранее, указывают на то, что мутации обоих генов приводят к сходным фенотипам.
Результаты
Все три пациента имели нормальный кариотип и были отрицательны в отношении хрупкой Х-мутации. Для трех пациентов был проведен анализ дозы всего генома с помощью 44K олигонуклеотидного массива CGH (G4426B Agilent Technologies, Пало-Альто, Калифорния). Патогенных вариантов не обнаружено.

Изображение в полном размере
Таблица в натуральную величину
обсуждение
Таблица в натуральную величину
Основываясь исключительно на клинических признаках, пациенты с мутациями в гене MED12 ранее были классифицированы по трем различным клиническим синдромам, хотя все они имеют в большей или меньшей степени общие клинические результаты: соматические нарушения роста (влияющие на вес и / или рост), измененный размер головы, микрогнатия, аномалии желчевыводящих путей, некоторые дисморфные черты лица, аномалии мочеполовой системы, аномалии рук и ног, в дополнение к проблемам в центральной нервной системе (агенез мозолистого тела, судороги, аутизм и / или гиперактивное поведение, умственная отсталость и задержка в развитии) (см. в [11, 18, 19]). Очень похожая картина была обнаружена среди пациентов с мутациями в гене MED13L, которые имеют широкий фенотипический спектр, который в основном включает те же клинические признаки, которые обнаруживаются в связи с мутациями в MED12 (13, 17, 20, 21).
Были описаны вредные варианты в генах других медиаторных комплексов: перицентрическая инверсия, вызывающая гаплоиндуктивность CDK19, связанная с двусторонними врожденными складками сетчатки, микроцефалией и легкой умственной отсталостью (23); Миссенс-варианты MED17 были связаны с постнатальной прогрессирующей микроцефалией с судорогами и атрофией головного мозга (24); Варианты MED23 вызывают аутосомно-рецессивный несиндромальный ID (25), а гомозиготный вариант MED25 вызывает аутосомно-рецессивное начало аксональной нейропатии Шарко-Мари-Тута у взрослых (CMT2B2, MIM # 605589) (26).
В дополнение к этим генам с мутациями, вызывающими заболевание, другие компоненты медиаторного комплекса являются хорошими генами-кандидатами для патологии. Ген MED1 был нарушен на мышиной модели, в которой нулевые мутанты погибли на ранней стадии гестации из-за сердечной недостаточности и обнаруживали нарушение развития нейронов с обширным апоптозом мозга (27). Гаплоиндуцированные мыши показали задержку роста, гипотиреоз гипофиза и широко нарушенную транскрипцию в определенных органах. С другой стороны, гомозиготная трансверсия в гене MED20 была обнаружена у двух пострадавших сестер из родственной австрийской семьи, у обеих из которых были обнаружены инфантильные дегенерации базальных ганглиев и атрофия головного мозга (28). Пациенты имели прогрессирующее нейродегенеративное расстройство, характеризующееся сильно задержанным психомоторным развитием, спастичностью и дистонией.
Анализы иммунопреципитации хроматина показали, что истощение MED26 приводило к снижению фосфорилирования С-концевого домена РНК pol II и уменьшению рекрутирования РНК pol II и компонентов SEC AFF4 и CDK9 как в промоторные области, так и в тела генов MYC и HSP70. (29). Авторы предположили, что MED26 может функционировать в качестве молекулярного переключателя, который в первом случае взаимодействует с инициирующим комплексом RNA pol II посредством прямых взаимодействий с TFIID, а на втором этапе обменивает TFIID с факторами удлинения RNA pol II для облегчения продуктивного удлинения. Мультибелковый комплекс TFIID состоит из TATA-связывающего белка (TBP) и 12 дополнительных TBP-ассоциированных факторов (TAFs) (30). Варианты, наблюдаемые в белках этого комплекса (TAF1, TAF2 и TBP), были вовлечены в несколько синдромальных форм умственной отсталости и задержки развития (31, 32, 33, 34), а также в X-сцепленной дистонии-паркинсонизме (35)., 36).
Kagey et al. (37) раскрыли, что Mediator и cohesin (SMC1A, MIM # 300040) физически и функционально связывают энхансеры и промоторы активных генов в мышиных эмбриональных стволовых клетках. Комплекс медиатора взаимодействует с когезином и образует кольцевой комплекс, который соединяет два сегмента ДНК, потому что между энхансерами и промоторами, занимаемыми медиатором и когезином, образуется петля ДНК. Фактор нагружения когезином NIPBL (MIM # 608667) связан с комплексами медиатор-когезин, обеспечивая механизм загрузки когезина в промоторы. Мутации в генах NIPBL и SMC1A вызывают синдром Корнелии де Ланге, генетически гетерогенный синдром, характеризующийся выраженными чертами лица, гирсутизмом, задержкой развития, умственной инвалидностью и аномалиями конечностей (38). Следует отметить, что фенотипический спектр синдрома Корнелии де Ланге включает в себя клинические признаки, которые широко распространены в синдромах МЭД, что также указывает на функционально связанную молекулярную этиологию. Некоторые фенотипические признаки, которые частично совпадают между синдромами МЭД и синдромом Корнелии де Ланге, включают опущенные ноздри, длинное пятно, высокое небо, низко посаженные уши, ухудшение слуха, косоглазие и умственную отсталость.
Клиническое сходство синдромов MEDs с другими генетическими состояниями, вызванными изменениями в генах, которые являются компонентами механизма инициации транскрипции, включая комплекс cohesin, комплекс TFIID или даже комплекс KMT2D, также примечательны. В целом, эти клинические сходства, вызванные различными генами, кодирующими функционально родственные белки, полностью согласуются с недавно предложенной концепцией «транскриптомопатий», класса нарушений, которые могут отражать глобальное нарушение регуляции транскрипции (40).
методы
Предметы
Письменное информированное согласие на исследование было получено от всех участников. Это исследование было рассмотрено и одобрено Комитетом по институциональной этике нашей больницы, и исследование было проведено в соответствии с Хельсинкской декларацией.
Фотографии пациентов, иллюстрирующие типичные черты лица и другие соответствующие клинические признаки пациентов 1 ( a и b ) с мутацией MED12 и пациентов 2 ( c и d ) и 3 ( e, f и g ) с мутациями MED13L.
Изображение в полном размере
Проектирование массива захвата, секвенирование следующего поколения и конвейер анализа
Специальная библиотека олигонуклеотидных зондов SureSelect была разработана для захвата 19 878 кодирующих экзонов из 614 патогенных и 642 генов-кандидатов, связанных с умственной отсталостью (рукопись готовится). Конструкция включает в себя все транскрипты, сообщенные для каждого целевого гена в разных базах данных (RefSeq, Ensembl, CCDS, Gencode, VEGA). Мастер проектирования стандартной ДНК SureSelect (Agilent Technologies) использовался для разработки зондов с плотностью укладки 2X и умеренно строгой маскировкой. Agilent Technologies (Санта-Клара, Калифорния) синтезировала в общей сложности 71 994 зонда, охватывающих 5, 073 Мбит / с (охват целевых объектов 99, 48%). Захват последовательности, обогащение и элюирование выполняли в соответствии с инструкциями производителя. Библиотеки секвенировали на платформе Illumina HiSeq 2000 с парным концом 2 × 90 б.п., следуя протоколу производителя для генерации по меньшей мере 100-кратной эффективной средней глубины.
Вызов вариации выполнялся на платформе DNAnexus (DNAnexus, Mountain View, CA) по следующему конвейеру: парные чтения Fasq были сопоставлены с эталонным геномом человека UCSC hg19 с использованием алгоритма BWA-MEM из пакета программного обеспечения BWA. Отображения были дедуплицированы с использованием Picard, выровнены по сайтам известных индилей, а их качество было перекалибровано путем просмотра ковариации в метриках качества с часто наблюдаемым изменением в геноме. После калибровки варианты вызывались с помощью модуля GATK Unified Genotyper. Этот конвейер следует рекомендациям Широкого института для лучших практик в варианте вызова. Были отфильтрованы варианты областей с низкой сопоставимостью или вариантов, в которых не было хотя бы одного образца с глубиной считывания ≥10. Аннотация вариантов нуклеотидов была выполнена с помощью программного обеспечения Ion Reporter (Life Technologies, Carlsbad, CA).
Приоритизация вариантов
Для оценки предполагаемого клинического влияния вариантов были применены следующие критерии: (i) частота аллелей



