Синдром фронтоназальной дисплазии. Возможности пренатальной диагностики и особенности медико-генетического консультирования


Фронтоназальная дисплазия (ФНД) – порок развития средней части лица, заключающийся в нарушении перемещения глаз по направлению к носу в процессе эмбриогенеза. Впервые был описан в 1967 г. W. De Myer [1]. Автор изучал больных с единообразным пороком развития и назвал это заболевание синдромом срединной расщелины лица. В 1970 г. S. Sedano и соавт. предложили переименовать этот синдром в синдром фронтоназальной дисплазии, так как расщелина лица не была облигатным признаком, строго определяющим этиопатогенез синдрома [2]. Этими же авторами был описан эмбриопатогенез ФНД: порок формируется с 19-го по 21-й день эмбрионального развития из-за нарушения миграции мезодермы, обусловленного мутацией в ALX3 гене [3].
Синдром ФНД характеризуется аутосомно-доминантным типом наследования с различной пенетрантностью (проявляемостью) и экспрессивностью (степенью выраженности). При этом чаще всего встречаются спорадические случаи, как проявление мутации de novo [4].
В OMIM (Online Mendelian Inheritance in Man) имеет номер #136760. В самой масштабной работе, посвященной изучению этого синдрома, проведенной в 1996 г., рассматривался 21 случай этой редкой патологии [5]. По данным авторов, соотношение больных мужчин и женщин составляет 2:1.
Манифестные (часто встречаемые) признаки синдрома ФНД или синдромальное «ядро» заболевания (рис. 1):
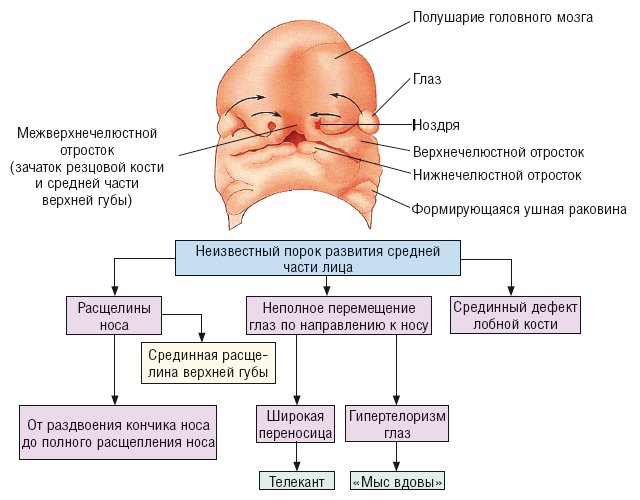
Рис. 1. Патогенез синдрома ФНД [6].
Глаза: узкие глазные щели, гипертелоризм, эпи-, телекант, катаракта, дегенерация сетчатки, колобома нижнего века.
Лоб: клиновидный рост волос на лбу («мыс вдовы»), срединный дефект лобной кости (скрытая расщелина черепа, лобное менингоэнцефалоцеле).
Нос: расщелины разной степени тяжести (от раздвоенного кончика носа до полной расщелины, возможно, в сочетании с широкой срединной расщелиной верхней губы), расщелины крыльев носа, широкая переносица, отсутствие кончика носа, назальные кожные привески.
Патология центральной нервной системы: агенезия мозолистого тела.
К редким симптомам, описанным при ФНД, относят: носовые и ушные привески, микрофтальм, низко расположенные уши, кондуктивную тугоухость, липомы на лбу и в мозолистом теле. В исследовании, посвященном синдрому ФНД [6], было установлено, что агенезия мозолистого тела встречалась в 57% случаев, липома мозолистого тела – в 19% случаев. Также описаны дефект межжелудочковой перегородки, тетрада Фалло, поли-, син-, брахидактилия, расщелина позвоночника, омфалоцеле, крипторхизм [7].
Прогноз для жизни и здоровья при синдроме ФНД зависит от наличия и тяжести сопутствующих аномалий. Пороки развития лица устраняются обычно серией пластических операций. По данным литературы [6, 7], интеллект у больных ФНД обычно сохранен. Однако W. De Myer отмечает умственную отсталость у 8% и легкое снижение интеллекта у 12% больных [1].
Пренатальная диагностика синдрома фронтоназальной дисплазии
Несмотря на то что изменения фенотипа при синдроме ФНД, казалось бы, очевидны: расщелина лица, агенезия мозолистого тела, патология мягких тканей носа, гипертелоризм и др. и не должны вызывать затруднений у врача ультразвуковой пренатальной диагностики, в литературе встречается ограниченное количество публикаций, посвященных пренатальной диагностике синдрома ФНД. Редкие работы о пренатальной диагностике единичных случаев синдрома посвящены в основном применению новых технологий 3D/4D с методиками поверхностной реконструкции [8–10]. Этот факт объясняется, скорее всего, тем, что врачи выставляют диагноз отдельных симптомов данного заболевания (чаще всего это расщелина губы/неба, лобная черепномозговая грыжа, агенезия мозолистого тела, патология развития мягких тканей носа) с перечислением всех найденных при ультразвуковом исследовании пороков без попытки провести клинико-синдромальный поиск. Такой «однобокий» подход к диагностике найденных аномалий не позволяет выставить правильный клинический диагноз синдрома ФНД, что в дальнейшем приведет к неполному и неадекватному медико-генетическому консультированию (МГК) семьи, которое заключается как в определении прогноза на данную беременность, так и в формировании тактики репродуктивного поведения семьи в дальнейшем и выработке специфических мер профилактики патологии.
Медико-генетическое консультирование при синдроме ФНД
При диагностике патологии с аутосомнодоминантным типом наследования изучение фенотипа/генотипа родителей позволяет установить, явилось ли данное заболевание следствием новой мутации (de novo) либо патологический ген унаследован от кого-то из родителей.
Если у одного из родителей находят даже малейшие признаки синдрома ФНД (учитывая различную пенетрантность и экспрессивность генов, которые определяют клиническую выраженность симптомов), риск повтора данной патологии составит 50%, в случае возникновения мутации de novo этот риск не превышает уровень общепопуляционного (1%), так как члены семьи здоровы.
Клиническое наблюдение 1
При проведении пренатальной эхографии в 34 нед беременности (настоящая беременность вторая, в семье один здоровый ребенок) в медико-генетическом отделении МОНИИАГ были выявлены лицевые дизморфии у плода женского пола – гипертелоризм, раздвоенный кончик широкого носа, образование в области переносицы (лобное менингоцеле малых размеров). Выставлен пренатальный диагноз синдрома ФНД, имеющей аутосомно-доминантный тип наследования, полностью подтвержденный после родов при осмотре новорожденного генетиком-синдромологом (рис. 2).
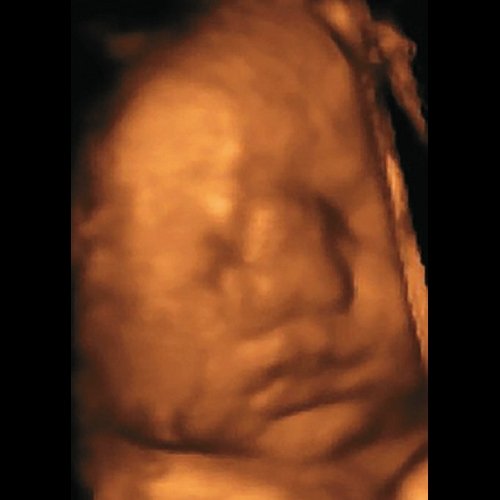
а) Пренатальный фенотип в 34 нед беременности.
Синдромы врожденных аномалий, проявляющихся карликовостью
Общая информация
Краткое описание
Протокол «Синдромы врожденных аномалий, проявляющихся карликовостью»
Код по МКБ:

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
1. Умственная отсталость.
2. Легкая умственная отсталость.
3. Умеренная умственная отсталость.
4. Тяжелая умственная отсталость.
5. Глубокая умственная отсталость.
Диагностика
Диагностические критерии
Жалобы и анамнез: задержка в психоречевом развитии, снижение мышления, памяти, внимания, расторможенность, врожденные пороки развития со стороны органов зрения, опорно-двигательного аппарата; отягощенная наследственность, перинатальная патология.
Физикальное обследование
Синдром Нунна: специфическое лицо с гипертелоризмом, антимонголоидным разрезом глаз, узкой и уменьшенной нижней челюстью, низко расположенными ушными раковинами. Шейная крыловидная складка или короткая шея, низкий рост.
Синдром Прадера-Вилли: ожирение, гипогенитализм, отставание в росте, мышечная гипотония. Заболевание проявляется уже у новорожденного с резко выраженной гипотонией. Мышечная гипотония является одним из ведущих диагностических признаков в раннем возрасте. На 2-4 году жизни гипотония заметно уменьшается, на первый план выступает булимия, развивается ожирение. В препубертатном возрасте или раньше может развиться инсулинрезистентный диабет. Умственная отсталость имеется практически у всех больных. Долихоцефалическая форма черепа.
Синдром Робинова-Сильвермена-Смита: необычное строение лица («лицо плода»), укорочение предплечий, гипоплазия половых органов, умеренная низкорослость. Типичными и постоянными аномалиями являются макроцефалия, выступающий лоб, широкая переносица, эпикант, гипоплазия средней части лица, короткий нос с вывернутыми вперед ноздрями, широкий фильтр, рот треугольной формы, гиперплазия десен. Скелетные аномалии заключаются в укорочении предплечий, брахидактилия, вывих бедра.
Лабораторные исследования: общий анализ крови и мочи.
Инструментальные исследования:
1. Электроэнцефалография (ЭЭГ): на ЭЭГ задержка формирования возрастной корковой ритмики, диффузные изменения электрогенеза головного мозга.
2. Электромиография (ЭМГ).
3. Компьютерная томография головного мозга (КТ).
Показания для консультации специалистов:
Публикации в СМИ
Дисплазия Арскога–Скотта
Дисплазия Арскога–Скотта — низкорослость, макроцитарная анемия, гемохроматоз, гепатомегалия, портальный цирроз, дефекты костей и суставов, стигмы дизэмбриогенеза. Генетические аспекты. Известны фенотипы: Â (100050, сильное влияние пола), À (*305400, большинство случаев), тип Кувейт (227330). Для фенотипа À (*305400, классический синдром) установлена локализация поражённого локуса (Xp11.21, ген FGD1 [FGDY, AAS]).
Клиническая картина. Задержка роста от лёгкой до умеренной, «мыс вдовы», гипоплазия верхней челюсти, нос с широкой спинкой и вывернутыми ноздрями, широкая верхняя губа, изогнутая линейная бороздка под нижней губой, гипертелоризм, птоз, антимонголоидный разрез глаз, косоглазие, астигматизм, увеличенная роговица, мягкие ушные раковины, расщелина верхней губы/нёба, шалевидная мошонка, крипторхизм, брахидактилия, контрактуры пальцев, клинодактилия, синдактилия, поперечная складка на ладони, лимфатический отёк стоп, слабость связок, рассекающие остеохондриты, переразгибание дистальных суставов пальцев, коленных суставов, плоскостопие, гиперподвижность шейных позвонков, макроцитарная анемия, гемохроматоз, гепатомегалия, портальный цирроз, неперфорированный задний проход, интерстициальные заболевания лёгких, деформация грудины.
Синонимы • Лице-генитальная дисплазия • Лице-генитальный синдром • Арскога–Скотта синдром • Лице-пальце-генитальная дисплазия • Лице-пальце-генитальный синдром
МКБ-10. Q87.5 Другие синдромы врождённых аномалий с другими изменениями скелета
Примечание. «Мыс вдовы» (194000, Â ) — линия роста волос на лбу в форме треугольника вершиной вниз, может наследоваться как доминантный признак, является компонентом ряда наследуемых синдромов (Арскога, Опитца–Фриаса, Варденбурга, фронто-назальной [136760] и кранио-фронто-назальной [304110] дисплазий и др.).
Код вставки на сайт
Дисплазия Арскога–Скотта
Дисплазия Арскога–Скотта — низкорослость, макроцитарная анемия, гемохроматоз, гепатомегалия, портальный цирроз, дефекты костей и суставов, стигмы дизэмбриогенеза. Генетические аспекты. Известны фенотипы: Â (100050, сильное влияние пола), À (*305400, большинство случаев), тип Кувейт (227330). Для фенотипа À (*305400, классический синдром) установлена локализация поражённого локуса (Xp11.21, ген FGD1 [FGDY, AAS]).
Клиническая картина. Задержка роста от лёгкой до умеренной, «мыс вдовы», гипоплазия верхней челюсти, нос с широкой спинкой и вывернутыми ноздрями, широкая верхняя губа, изогнутая линейная бороздка под нижней губой, гипертелоризм, птоз, антимонголоидный разрез глаз, косоглазие, астигматизм, увеличенная роговица, мягкие ушные раковины, расщелина верхней губы/нёба, шалевидная мошонка, крипторхизм, брахидактилия, контрактуры пальцев, клинодактилия, синдактилия, поперечная складка на ладони, лимфатический отёк стоп, слабость связок, рассекающие остеохондриты, переразгибание дистальных суставов пальцев, коленных суставов, плоскостопие, гиперподвижность шейных позвонков, макроцитарная анемия, гемохроматоз, гепатомегалия, портальный цирроз, неперфорированный задний проход, интерстициальные заболевания лёгких, деформация грудины.
Синонимы • Лице-генитальная дисплазия • Лице-генитальный синдром • Арскога–Скотта синдром • Лице-пальце-генитальная дисплазия • Лице-пальце-генитальный синдром
МКБ-10. Q87.5 Другие синдромы врождённых аномалий с другими изменениями скелета
Примечание. «Мыс вдовы» (194000, Â ) — линия роста волос на лбу в форме треугольника вершиной вниз, может наследоваться как доминантный признак, является компонентом ряда наследуемых синдромов (Арскога, Опитца–Фриаса, Варденбурга, фронто-назальной [136760] и кранио-фронто-назальной [304110] дисплазий и др.).
Краниостеноз у детей
О заболевании

Среди деформаций черепа встречаются врожденные и приобретенные. Врожденные появляются еще во время беременности, и ребенок рождается с определенным пороком развития. Приобретенные возникают после родов, чаще всего в результате травмы или определенного вмешательства, например, хирургического.
Краниостеноз у детей может быть как врожденным, так и приобретенным после рождения. Этот дефект представляет собой досрочное сращение или отсутствие швов в костях черепа, которые в норме должны оставаться пластичными для естественного роста и развития мозга ребенка. В результате внутричерепное давление повышается, а форма черепа видоизменяется. Если речь идет о пороке развития, краниостеноз у новорожденных может сопровождаться и другими дефектами, которые затрагивают головной мозг и другие органы тела. В случае с приобретенным заболеванием сращение швов происходит из-за травмы или хирургического вмешательства.
Краниостеноз встречается у одного из 1000 новорожденных.
Из-за чего возникает заболевание
Врачи единогласны во мнении: наследственность на преждевременное сращение костей черепа не влияет. И если с приобретенным заболеванием ситуация более или менее понятна, то на появление врожденного с разной долей вероятности могут влиять:
Чем опасен краниостеноз
Отсутствие подвижных швов в черепе остро ограничивает головной мозг. Поэтому чем раньше приходит помощь при краниостенозе, тем более высока вероятность полного излечения ребенка в будущем. Механизмы компенсации у младенцев до 2 лет очень высоки, реабилитация же после этого возраста оказывается более сложной и продолжительной. Если не пройти лечение своевременно, заболевание может вызывать следующие состояния:
Если же не пройти лечение своевременно, заболевание может вызывать следующие состояния:
Каким бывает краниостеноз у детей
В первую очередь зависит от того, есть ли у ребенка другие пороки развития. Иногда патология сопровождает различные синдромы — тогда ее называют синдромальной. Например, она может возникать одновременно со сращением пальцев на руках или ногах, расщелиной нёба или губы, мозговыми грыжами.
Если сращение швов в костях черепа происходит без других дефектов развития, оно считается несиндромальным, то есть самостоятельным.
Врачи классифицируют болезнь на виды исходя из того, какие именно черепные швы срастаются:
Синостоз — или сращение швов — может вовлекать в себя от одного до нескольких швов. Существует понятие пансиностоза — полного зарастания всех швов. Такой вид патологии можно считать наиболее тяжелым, он встречается реже остальных.
Как проходит лечение краниостеноза у детей
Способ устранить патологию только один — хирургическая операция. Ее проводят, чтобы восстановить форму костей черепа. После того, как кости принимают естественную форму, их скрепляют мини-шурупами или мини-пластинами, хирургической проволокой, которые удаляют через год. Их удаляют через год. В ходе операции врачи используют современный технологичный биодеградирующий материал, который не нужно удалять хирургически — он постепенно растворяется сам, а на его месте прирастает собственная костная ткань. Это существенно упрощает восстановление пациента.
Наилучших результатов можно ожидать, если операция проводится в возрасте от 3–4 месяцев до 2 лет. Сильные деформации черепа иногда можно заметить только после того, как кости заканчивают свое формирование, то есть в возрасте 5–6 лет. В таких случаях патология проявляет себя сильными головными болями, ухудшением зрения и повышением внутриглазного давления, повышенной утомляемостью и раздражительностью.
Если заболевание возникает как ответ на трепанацию черепа или после травмы, деформированный участок кости удаляют, а на его место устанавливают имплант из современного материала — полимера, металла или керамики.
Медицинские организации
Помощь благотворительного фонда «Линия жизни» в лечении детей с краниостенозом заключается в покупке перевязочных материалов и инструментов, которые требуются для коррекции дефекта.
Новый взгляд на патогенез и лечение андрогенетической алопеции
Евгений КАРАСЕВ, к. м. н., врач-трихолог, Москва
Татьяна ВИННИК, PhD, врач-дерматовенеролог, трихолог, Астана
Андрогенетическая алопеция (АГА) – сложное мультифакториальное состояние, основным проявлением которого являются истончение и поредение волос в лобно-теменной зоне скальпа как у мужчин, так и у женщин.
АГА развивается с годами и является отражением сегментарного или органоспецифичного преждевременного старения. Рассматривается как неизбежное следствие, так как генетически обусловлена и проявляется у любого человека, как правило, после 30 лет.
«Виновным» геном в данном случае считается ген андрогенного рецептора (AR), который находится на хромосоме X. «Сидит» он латентно, никак себя не проявляя до поры до времени. У каждого индивидуума эта пора своя. Однако, если взглянуть в целом на мужскую половину человечества, в зависимости от активности этого гена их можно разделить на две довольно большие группы: мужчины с высокой активностью АГА-гена и мужчины с невысокой активностью того же гена. У представителей первой группы первые признаки облысения регистрируются в 18–20 лет, когда на лбу появляются ползущие на темя «андрогенетические заливы», формирующие примерно к 30–35 годам отчетливо проглядывающую букву «М». У представителей второй группы происходит то же самое (только с меньшей скоростью!), начинает формироваться примерно в 45 лет. Но финал у всех более или менее одинаковый – лобно-теменное облысение, если не принимать специальных лечебно-профилактических мер. Эволюция клинической картины развивается по сценарию, предложенному в 70-х годах прошлого века Норвудом–Гамильтоном.
Итак, при АГА у мужчин никогда не редуцируются только волосы, образующие узкую кайму на висках и затылке.
Что же касается женской половины человечества, то она в зависимости от активности данного гена распадается на три довольно большие группы: лица с высокой активностью АГА-гена, со средней и с невысокой активностью того же гена. У представительниц первой группы первые признаки облысения регистрируются в 18–20 лет, когда в лобно-теменной зоне начинается постадийный процесс, описанный в 1977 году Людвигом (рис. 2).
У представительниц второй группы то же самое, только с меньшей скоростью, начинает формироваться в 35–40, а у третьей – при наступлении менопаузы, то есть в 50–55 лет.
В редких случаях чрезвычайно тяжелая АГА или ее раннее начало может быть симптомом сложного генетического заболевания, например трихоринофалангеального синдрома, прогерии, синдрома Ларона, миотонической дистрофии Куршмана – Штейнерта – Баттена и пр.
ПРИЧИНЫ РАЗВИТИЯ АГА
В основе развития АГА – генетически обусловленные особенности метаболизма андрогенов в волосяном фолликуле.
Наиболее значимым фактором у мужчин является повышенная активность фермента 5α-редуктазы II типа, которая в генетически предрасположенных волосяных фолликулах метаболизируют тестикулярный тестостерон в дигидротестостерон (ДГТ).
Помимо аналогичных с мужскими локальных метаморфоз андрогенов у женщин в патогенезе АГА, как правило, также большую роль играет снижение активности ароматазы, которая преобразует
циркулирующий в крови тестостерон яичников в 17 бета-эстрадиол.
Увеличение локальной концентрации ДГТ приводит к прогрессирующему сокращению анагена за счет более длительной фазы телогена (рис. 3) и сопровождается прогрессирующей миниатюризацией волосяных фолликулов. Последняя осуществляется за счет относительно резких сокращений количества клеток дермального сосочка и дермальной оболочки.
В дополнение к андрогензависимым изменениям в патогенезе АГА доказано вовлечение фолликулярного микровоспаления с формированием фиброза, спровоцированного присутствующей бактериальной флорой, токсинами и окислительным стрессом.

МЕТОДЫ ЛЕЧЕНИЯ
Лечение АГА прежде всего нацелено на увеличение волосяного покрова кожи головы и предотвращение истончения и поредения волос в будущем. На ранних стадиях оно эффективнее, поскольку изменения волосяных фолликулов не носят необратимого характера.
Существуют как терапевтические, так и хирургические методы восстановления роста волос при АГА: прием модификаторов биологических реакций, гормональных и негормональных антиандрогенов, блокаторов 5α-редуктазы и трансплантация волос.
Так, примером патогенетических средств лечения АГА являются препараты растительного происхождения, получаемые из листьев оливкового дерева, вытяжки из корня лопуха, крапивы двудомной, у женщин – некоторые оральные контрацептивы и спиронолактон.
Радикальным методом восстановления волос при АГА является трансплантация собственных волосяных фолликулов. Принцип хирургического лечения заключается в перемещении андрогеннезависимых терминальных волосяных фолликулов из не подверженной облысению затылочной зоны в участки андрогензависимого поредения. Пересадка волос – это успешный метод лечения АГА с долговременным эффектом. Тем не менее естественное прогрессирование облысения будет продолжаться, и могут потребоваться последующие пересадки, чтобы трансплантированный участок не оказался окруженным кожей, лишенной волос.
Среди наружных методов коррекции АГА упомянем классический миноксидил и ставшие популярными в последнее десятилетие трехфазные комплексы, содержащие «золотую троицу»: вазодилататор эпигенин, трипептидный фактор роста волос и олеаноловую кислоту. Последняя особенно важна при АГА, так как ингибирует 5α-редуктазу. Применяются эти средства по разным схемам в зависимости от стадии процесса. Одним из наиболее терапевтически успешных представителей этого класса препаратов является ДЕКОПИЛЛ™/DEKOPILL™ от медицинской компании Charismo (Даллас, США).
ЭФФЕКТИВНОЕ СРЕДСТВО КОРРЕКЦИИ АЛОПЕЦИЙ
ДЕКОПИЛЛ™ – это новейший запатентованный натуральный биоактивный пептидный комплекс с витаминами, аминокислотами и озонидами, предназначенный для замедления процесса патологического выпадения волос, прошедший испытания и показавший клинически значимые результаты защиты и восстановления фолликулов, а также улучшения структуры и здоровья волоса.
ДЕКОПИЛЛ™ предназначен для устранения основных симптомов патологической утраты волос большинства известных алопеций. Основной результат его действия заключается в продлении фазы роста волоса и улучшении устойчивости к вредному воздействию ДГТ, а также в увеличении кровообращения, усилении витаминизированного питания корней волос и поверхности кожи головы, вследствие чего замедляется процесс старения фолликулов.
Создатели препарата потратили 10 лет на разработку и клинические испытания, которые показали, что эффекты коррекции алопеций достоверно регистрируются через 30–90 дней от момента начала комплексного лечения.
Быстрое уменьшение количества выпадающих волос (после 30-дневного курса).
Активизация латентно существующих волос (от 0 до 3 мм после 90-дневного курса).
АКТИВНЫЕ КОМПОНЕНТЫ ПРЕПАРАТА:
Доказано, что для эффективного лечебного воздействия при алопециях необходимо стимулировать кровообращение кожи головы не менее 8 часов в сутки. (Для сравнения: перец, горчица, имбирь стимулируют кровообращение не более 1 часа!) Именно этот эффект в препарате достигается с помощью эпигенина неэфиромасличного генеза, что предотвращает любые аллергические реакции.
Возможны покраснения (так как происходит поверхностное расширение сосудов) при протекании на кожу лица или утром при споласкивании волос, которые исчезают через 5–10 минут.
Противопоказания: беременность, онкология в активной форме, частые эпилептические припадки.
ОСНОВНЫЕ ТЕРАПЕВТИЧЕСКИЕ ЭФФЕКТЫ ПРЕПАРАТА:
СПОСОБ ПРИМЕНЕНИЯ ПРЕПАРАТА
Каждый вечер наносите от 5 до 9 полных пипеток лосьона на сухую поверхность кожи головы, мягко промассируйте голову и затем вымойте руки. Оставьте нанесенный лосьон как минимум на 8 часов. На следующее утро допустимо мытье или ополаскивание головы водой.
Минимальный курс применения – 30 дней, наиболее часто рекомендуемый курс применения – 90 дней.
1 упаковка рассчитана на 30-дневный курс – 120 мл (4 флакона х 30 мл).
Для наиболее быстрого и стойкого лечебного эффекта возможно применение лосьона ДЕКОПИЛЛ™ с другими терапевтическими методами:
ЭПИЛОГ
В заключение хотелось бы сделать особый акцент на двух аспектах.
Во-первых, применение всех вышеперечисленных методов лечения не меняет генетическую программу волосяных фолликулов, поэтому терапевтические успехи данных мероприятий носят временный характер и их следует регулярно повторять в течение всей жизни пациента. Также необходимо исключать внутренние факторы (например, железодефицитную анемию, эндокринные заболевания и пр.), совместно с АГА ухудшающие состояние волос и приводящие к их хроническому диффузному поредению.
Во-вторых, для скептиков отметим, что инновационный ДЕКОПИЛЛ прошел строгий дерматологический контроль в клиниках Европы и США, показав при этом свою эффективность и безопасность в результате клинических тестов, которые объективно выявили:



