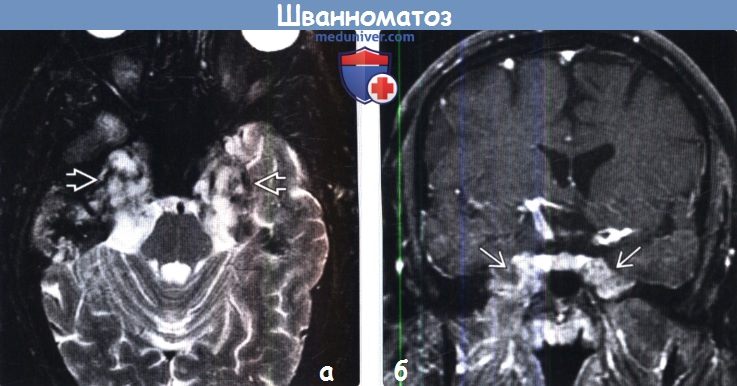
Шванноматоз
Шванноматоз – это редкая форма нейрофиброматоза, которая была выявлена не так давно, характеризующаяся образованием множественных шванном, но несоответствующая критериям нейрофиброматоза 2 типа. Шванноматоз связан с мутацией гена SMARCB1 на 22 хромосоме. вызывает развитие доброкачественных опухолей — шванном — обычно на спинном и периферических нервах.
По данным американской ассоциации CTF каждый день в мире рождается 9 человек со шванноматозом. Это значит, что новый пациент со шванноматозом рождается каждые 3 часа.
Симптомы шванноматоза обычно возникают в возрасте от 30 до 60 лет, хотя они могут возникнуть в любом возрасте. Основным клиническим проявлением шванноматоза является развитие шванном, отличительных опухолей, которые растут на периферических нервах. Наиболее распространенным признаком является хроническая боль. Считается, что это вызвано, по крайней мере, частично шванномами, которые разрастаясь давят на нервы. В некоторых случаях боль, которую испытывают люди, непропорциональна размеру имеющихся опухолей. Кроме того, интенсивность и частота боли значительно варьируется среди пациентов. У многих пациентов со шванноматозом боль является единственным симптомом заболевания, по этой причине шванноматоз часто трудно диагностировать. Самое важное отличие от нейрофиброматоза 2 типа заключается в том, что у пациентов со шванноматозом обычно не развиваются опухоли на вестибулярном нерве, которые вызывают потерю слуха у людей с НФ2 (хотя редкие случаи были зарегистрированы). Кроме того, другие типы опухолей, которые могут возникнуть у людей с НФ2 (включая менингиомы, эпендимомы и астроцитомы), не встречаются у тех, кто болен шванноматозом, за редким исключением. Особенности НФ1, такие как проблемы с обучением и пятна цвета “кофе с молоком”, также не присутствуют у людей со шванноматозом.
Опухоли шваннома
Шваннома является новообразованием доброкачественной природы, развивающимся из шванновских клеток. Последние формируют миелиновую оболочку нервных волокон и по неустановленным причинам способны начать слишком активное размножение, которое и приводит к появлению опухоли. Шваннома поражает разные нервы, однако чаще всего её диагностируют на слуховом. На её долю приходится 10% случаев церебральных, 20% спинномозговых и 50% опухолей периферических нервов.
Пройти хирургическое лечение шванномы в Москве приглашает отделение нейрохирургии ЦЭЛТ. Уже более 30-ти лет мы работаем на отечественном рынке платных медицинских услуг и располагаем всем необходимым для проведения лечения в соответствии с международными стандартами. Наши нейрохирурги имеют за плечами десятилетия опыта практической и научной работы и применяют эффективные щадящие методики.
Этиология шванномы
Несмотря на то, что учёным удалось установить связь между развитием шванномы и чрезмерно активным размножением шванновских клеток, причина последнего до сих пор не известна. Существует ряд теорий, согласно которым инициирующие факторы развития опухоли — следующие:
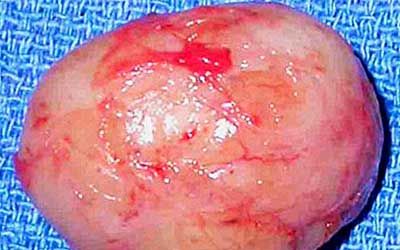
Новообразование растёт медленно и имеет вид круглого или овального узла с неровной поверхностью, покрытой капсулой. Внутри шванномы нередко обнаруживаются кисты разного размера, заполненные патологической жидкостью.
Клиника шванномы
Из-за медленного роста шванномы и практически полного отсутствия клинических проявлений, её обнаруживают достаточно поздно. Это происходит на том этапе, когда она начинает сдавливать соседние ткани, а поражённый нерв теряет способность нормально выполнять свои функции. Клиническая картина напрямую зависит от того, какой именно нерв повреждён:
Осложнения шванномы
Как уже упоминалось, часто шванному обнаруживают когда она уже выросла до внушительных размеров. Раннее диагностирование происходит, как правило, случайно: если пациент обращается к врачу по поводу другой проблемы. Своевременность лечения играет важную роль, поскольку позволяет исключить следующие осложнения:
Наши врачи



Диагностика и лечение шванномы в ЦЭЛТ
В нейрохирургии ЦЭЛТ работают кандидаты и доктора наук, врачи высшей категории, проводящие научную и практическую работы. Они располагают мощной диагностической базой, позволяющей точно ставить диагноз. Перед тем, как разработать тактику лечения шванномы, они проводят комплексную диагностику, позволяющая установить её размер, локализацию, влияние на окружающие ткани, кистозные изменения. Для этого назначают:
Пройти полное обследование и начать лечение можно в многофункциональной клинике ЦЭЛТ – последние достижения медицины на страже вашего здоровья.
Хотите узнать больше? Интересует стоимость удаления шванномы? Записывайтесь на приём к нашим специалистам и получайте ответы на любые вопросы: +7 (495) 788-33-88.
При каком наследственном заболевании встречается множественный шванноматоз
а) Определение:
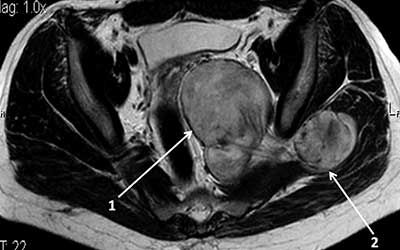
• Шванноматоз: множественные шванномы периферической нервной системы без вовлечения вестибулярных нервов
1. Общие характеристики шванноматоза:
• Лучший диагностический критерий:
о Множественные инкапсулироанные отграниченные объемные образования, расположенные вдоль хода черепных и периферических нервов без вовлечения ЧН VIII
2. КТ при шванноматозе:
• Бесконтрастная КТ:
о Образования плотностью от изо- до слегка гиподенсных по отношению к мозговой ткани
о Ищите влияние образования на прилежащие кости и отверстия
— Равномерное увеличение:
— Тонкие, склеротические края («хирургические»)
• КТ с контрастированием:
о Вариабельное, часто гетерогенное контрастирование
 (а) КТ области шеи с контрастированием, аксиальный срез: у женщины 31 года с шванноматозом в надскладочной области гортани определяется инкапсулированное гетерогенно контрасти-руемое объемное образование с четкой границей, почти полностью сдавливающее просвет гортани.
(а) КТ области шеи с контрастированием, аксиальный срез: у женщины 31 года с шванноматозом в надскладочной области гортани определяется инкапсулированное гетерогенно контрасти-руемое объемное образование с четкой границей, почти полностью сдавливающее просвет гортани.
(б) КТ с контрастированием, корональная реконструкция: определяются дополнительные объемные образования гетерогенной плотности, распространяющиеся на левое плечевое сплетение и яремное отверстие. Описанные образования являются множественными шванномами у данного пациента со шванноматозом.
3. МРТ при шванноматозе:
• Вариабельная интенсивность сигнала на всех последовательностях вследствие различного количества областей Антони А и Антони В
• Гиперинтенсивный сигнал на Т2-ВИ, PD-ВИ, FLAIR и STIR
• Интенсивное, обычно гетерогенное контрастирование на постконтрастных Т1-ВИ
4. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о Постконтрастная МРТ является золотым стандартом диагностической визуализации шванноматоза
в) Патология шванноматоза:
1. Общие характеристики:
• Генетика:
о Сложные не до конца понятые генетические особенности, включающие вовлечение гена SMARCB1 в клетках зародышевой линии и НЕ обнаруживающиеся при спорадических, несиндромных шванномах:
— Ген супрессора опухоли, расположенный в 22 хромосоме
— Вероятно наличие феномена множественных мутаций с мутациями генов SMARCB 1 и NF2 в пораженных тканях
г) Клиническая картина шванноматоза:
1. Проявления:
• Наиболее частые признаки/симптомы:
о Боль, обычно невропатического характера; может приводить к потере трудоспособности:
— В отличие от НФ2, чаще проявляющегося неврологической недостаточностью
о Симптомы обычно развиваются на втором или третьем десятилетии жизни:
— В отличие от НФ1 (первое десятилетие) и НФ2 (второе десятилетие)
2. Демография:
• Эпидемиология:
о Сведения о встречаемости варьируют от 1/40000 до 1/1,7 миллиона, возможно ближе к 1/40000 (сходно с НФ2)
3. Течение и прогноз:
• НОРМАЛЬНАЯ продолжительность жизни в отличие от пациентов с НФ2, у которых ожидаема сокращенная продолжительность жизни
4. Лечение:
• Контроль симптомов путем контроля над болью
• Хирургическое вмешательство только при компрессии спинного мозга или симптомах, несомненно обусловленных шванномой
• Отсутствие роли лучевой терапии, развитие роли химиотерапии
д) Диагностическая памятка:
1. Обратите внимание:
• У пациента может быть шванноматоз при о Возрасте > 30:
о Наличии > одной шванномы
о Отсутствии шванном слухового нерва
2. Советы по интерпретации изображений:
• НЕ следует отдавать предпочтение дигнозу шванноматоз у пациента моложе 30 лет, поскольку шваннома слухового нерва может развиться позже:
о С ↑ возраста пациента ↑ вероятность шванноматоза и ↓ вероятность НФ2
3. Советы по отчетности:
• При обследовании пациента >30 лет с наличием множественных невестибулярных шванном рентгенолог должен как предполагать шванноматоз, так и рекомендовать проведение высокоразрешающей МРТ височных костей/ВСП для исключения НФ2
е) Список литературы:
— Вернуться в оглавление раздела «Лучевая медицина»
Редактор: Искандер Милевски. Дата публикации: 5.3.2019
Содержание
Дерматология в России
Зарегистрируйтесь!
Если Вы врач, то после регистрации на сайте Вы получите доступ к специальной информации.
Если Вы уже зарегистрированы, введите имя и пароль (форма в верхнем правом углу или здесь).
Нейрофиброматоз: обзор, посвящённый NF1, NF2 и шванноматозу
Нейрофиброматоз: обзор, посвящённый NF1, NF2 и шванноматозу
Нейрофиброматоз – гетерогенная группа наследственных онкологических синдромов, приводящих к опухолям центральной и периферической нервной систем.
На сегодняшний день наиболее распространенной формой является нейрофиброматоз 1-го типа (NF1, 96%), за которым следует нейрофиброматоз 2-го типа (NF2, 3%) и менее известная форма – шванноматоз. Нейрофиброматоз не имеет половой или расовой предрасположенности.
Нейрофиброматоз 1 типа
Введение. NF1, также известный как болезнь фон Реклингхаузена или периферический нейрофиброматоз, является синдромом аутосомно-доминантной предрасположенности к опухоли, наиболее часто характеризующимся развитием множественных нейрофибром периферических нервов.
Ожидаемая продолжительность жизни сокращается в среднем до 54 лет, часто из-за злокачественных новообразований (2).
Злокачественные новообразования, связанные с NF1, включают злокачественные опухоли оболочек периферических нервов, глиомы, лейкемию, феохромоцитомы, стромальные опухоли желудочно-кишечного тракта ( GI ) и другие. NF1 вызван мутацией в гене-супрессоре опухоли нейрофибромина, расположенном на хромосоме 17.
Он имеет высокую пенетрантность, а частота мутаций гена NF высока, причем 80% имеют наследование по линии отца (3). Самое раннее известное изображение предполагаемого нейрофиброматоза датируется 13 веком с рисунками цистерцианского монаха (4).
В 1862 году Вирхов сослался на наследственный компонент, когда описал человека, у которого “тело было полностью покрыто шишками размером от булавочной головки до голубиного яйца”, и он отметил, что “проявления наследовались уже в течение трех поколений».
Однако именно студент Вирхова Фридрих фон Реклингхаузен в 1882 году дал наиболее полное клиническое и гистологическое описание этого заболевания и ввел термин “нейрофиброма”.
Диагностические критерии. Симптомы NF1 могут значительно различаться у пациентов, и поэтому своевременный и конкретный диагноз трудно установить.
Хотя клинический диагноз может быть заподозрен очень рано в детстве или младенчестве, основные признаки могут полностью отсутствовать до более старшего возраста.
Приблизительно 30% пациентов с NF1 будут соответствовать одному из приведенных ниже критериев к возрасту 1 год, 97% пациентов будут соответствовать двум критериям к возрасту 8 лет; в ретроспективном обзоре пациентов с NF1, все пациенты соответствовали критериям к возрасту 20 лет (1).
В 1987 году Национальные институты здравоохранения (NIH) США разработали консенсус в отношении диагноза из-за клинических различий.
Клинический диагноз может быть установлен, если критерии, перечисленные в таблице 1, удовлетворяются без возможного альтернативного диагноза.
Патогенез и генетические особенности. Ген, ассоциированный с NF1, был идентифицирован в 1990 году, и было обнаружено, что он является одним из крупнейших генов в геноме человека, охватывающим 280 kbp ДНК (6). Ген NF1 расположен на хромосоме 17q11.2, которая кодирует белок, известный как нейрофибромин (7).
Нейрофибромин относится к активирующему GTPase семейству белков-супрессоров опухоли, которые регулируют функцию передачи сигналов RAS/MAPK и механистическую мишень рапамицина (mTOR). Примерно 50% мутаций происходят de novo у пациентов без семейного анамнеза.
Для семей с унаследованной мутацией наблюдается полная пенетрантность; однако клинические проявления у членов семьи могут сильно различаться. Вариабельность фенотипической экспрессии, вероятно, является результатом эпигенетической модификации (8).
Исследования, посвященные корреляции генотипа и фенотипа, показали, что делеция всего гена, известная как микроделеция 17q11.2, связана с более тяжелой формой заболевания, в то время как мозаицизм может привести к легкому или даже сегментарному изменению (6, 7).
Было идентифицировано почти 1500 различных мутаций гена NF1, и эти мутации были обнаружены по всей длине большого локуса гена (6).
Нонсенс, сдвиг рамки считывания и точечные мутации были идентифицированы с большинством мутаций, приводящих к укороченной форме белка.
Генетическое тестирование часто проводится у членов семьи пациента, у которого была выявлена патологическая мутация, и, таким образом, конкретная мутация может быть проверена у их родственников.
В противном случае, учитывая удивительно высокую степень подверженности мутациям и отсутствие “горячих точек” на гене, NF1 не поддается мутационному анализу в качестве практического диагностического инструмента (6).
Диагноз NF1 чаще всего ставится на основании клинических данных, рассмотренных выше.
Клинические проявления
Нейрофибромы. Нейрофибромы являются наиболее распространенным типом опухолей NF1, встречающейся примерно у 60% пациентов. Гистологически нейрофибромы NF1 неотличимы от спорадических опухолей, хотя первые часто бывают крупнее.
Нейрофибромы могут быть кожными или внутренними, затрагивающими глубокие мягкие ткани. Кожные формы могут быть пятнистыми, узловатыми или бляшкообразными, развивающимися в позднем детстве и увеличивающимися в количестве во взрослом возрасте (2).
Внутренние или глубокие нейрофибромы могут возникать по всему телу, включая периорбитальные, забрюшинные области, желудочно-кишечный тракт и средостение (9).
Патогномоничные для NF-1 плексиформные нейрофибромы – это внутренние нейрофибромы, которые вместо того, чтобы расти интраневрально в пределах одного нерва, разрастаются, вовлекая несколько пучков или ветвей нерва или сплетения.
Эта модель роста соответствует характерному описанию “мешка с червями”, данному при пальпации или хирургическом исследовании этих опухолей. Плексиформные нейрофибромы часто развиваются в детстве и быстро растут, оказывая массовое воздействие на соседние структуры.
В отличие от кожной формы, плексиформные нейрофибромы имеют повышенный риск трансформации в злокачественные опухоли оболочек периферических нервов (MPNST). MPNST являются редкими агрессивными веретеноклеточными саркомами, на долю которых приходится лишь около 5% всех сарком мягких тканей.
Около 50% MPNST развивается при NF1, поскольку у этих пациентов риск развития одного из них в течение жизни составляет от 8 до 13% (3).
Внезапное изменение или рост плексиформной нейрофибромы при наблюдении за визуализацией, а также повышенное поглощение фтордезоксиглюкозо-позитронно-эмиссионной томографии (ПЭТ-сканирование) должны вызывать подозрение на злокачественную трансформацию.
Лечение нейрофибром включает хирургическую резекцию и лазерную терапию, в то время как цель хирургии плексиформной нейрофибромы часто заключается в ее удалении. Недавние клинические испытания с участием ингибитора тирозинкиназы иматиниба показали уменьшение объема опухоли более чем на 20% у подгруппы пациентов (10).
В настоящее время проводятся дальнейшие исследования, посвященные этой терапии, а также ингибиторам mTOR (10).
MPNST являются значительной причиной смертности у пациентов с NF1, и, несмотря на радикальное иссечение с широким хирургическим доступом с последующим химиолучевым лечением, 5-летняя выживаемость остается низкой из-за частых метастазов в легких и костях, а также местных рецидивов (2).
Пигментные аномалии. Пятна кофе с молоком – это доброкачественные коричнево-бежевые пигментированные макулы, которые могут встречаться в любом месте тела и часто являются признаком NF1. К возрасту 1 года у 99% пациентов с диагнозом NF1 будет шесть или более макул цвета кофе с молоком размером более 5 мм (препубертатные критерии в соответствии с NIH) (1, 9).
Хотя пятна с кофе с молоком являются общей особенностью NF1, они неспецифичны, поскольку их можно увидеть примерно у 10% населения в целом, а также при других генетических синдромах – карликовости Сильвера Рассела, MEN IIb, синдроме Легиуса и синдроме Маккьюна-Олбрайта.
Количественное ограничение, используемое в диагностических критериях, основано на исследовании Кроу и др. в 1956 году, в котором 78% из 203 проанализированных пациентов с NF1 имели по крайней мере шесть пятен от кофе с молоком размером более 15 мм (11).
Это количество больше, чем указано для населения в целом. В ретроспективном обзоре младенцев с родимыми пятнами Mihm и соавт. показали, что у 1,8% афроамериканских младенцев будет три или более пятен цвета кофе с молоком, в то время как у кавказских младенцев редко бывает два или более (12).
Гистологически эти высыпания представляют гиперпигментацию базального эпидермиса с присутствием макромеланосом (9).
Хотя возможности для злокачественной трансформации нет, иногда требуется косметическое лечение.
Отчеты о случаях показывают, что у некоторых пациентов может быть хороший ответ на дерматологическую лазерную терапию для депигментации; однако для получения ответа может потребоваться несколько процедур, и у части пациентов рецидив пигментации произойдет в течение 6 месяцев (13).
Пигментные пятна в подмышечной впадине и паху являются еще одним определяющим симптомом NF1. Только примерно 40% пациентов будут иметь пигментные пятна в младенчестве, а у 90% пациентов с NF1 они появятся к 7 годам (1).
Меланоцитарные узелки также могут встречаться в радужной оболочке у пациентов с NF1. Эти небольшие, часто множественные гамартоматозные поражения, известные как узелки Лиша, встречаются у 93% взрослых с NF1. Они протекают бессимптомно (14).
Глиомы. Пациенты с NF1 подвергаются повышенному риску развития глиом низкой и высокой степени злокачественности. Наиболее часто встречающейся глиомой в условиях NF1 является глиома зрительного нерва низкой степени злокачественности.
Эти глиомы зрительного нерва встречаются примерно у 15% пациентов с NF1 и обычно появляются в возрасте 7 лет (2).
Как правило, эти опухоли являются пилоцитарными астроцитомами I степени по классификации Всемирной организации здравоохранения и гистологически эквивалентны пилоцитарным астроцитомам общей популяции с двухфазным ростом, волосоподобными отростками и волокнами Розенталя.
Учитывая вялотекущий характер роста этих опухолей, лечение обычно состоит из наблюдения. Когда острота зрения снижается, может быть применена химиотерапия (2).
Расположение этих опухолей не позволяет проводить хирургическое вмешательство, и у пациентов с NF1 лучевой терапии избегают из-за повышенного риска развития злокачественных новообразований, вызванных радиацией (15).
Глиомы ствола головного мозга являются второй по частоте глиомой NF1, и опять же, эти опухоли обычно являются пилоцитарными астроцитомами.
По тем же причинам, что и при глиомах зрительного нерва, химиотерапия является единственным доступным методом лечения с целью уменьшения симптомов и повышения длительности выживания.
Наконец, пациенты с NF1 имеют пятикратный риск развития злокачественных глиом, особенно глиобластом, по сравнению с общей популяцией со средней выживаемостью около 1 года.
Скелетно-мышечные нарушения. У детей с NF1 значительно повышен риск развития рабдомиосаркомы, что примерно в 20 раз выше, чем у населения в целом (2).
Эти рабдомиосаркомы могут возникать в любом месте; однако систематический обзор показал преобладание локализации в тканях мочевого пузыря и предстательной железы (16). Протоколы лечения, применяемые при несиндромных рабдомиосаркомах, также могут быть применены к пациентам с NF1.
У пациентов с NF1 часто отмечаются различные аномалии скелета, включая остеопению, сколиоз или врожденную дисплазию большеберцовой кости. Кроме того, многие пациенты с NF1 невысокого роста, хотя пропорции их тела остаются нормальными (17).
Механистическая связь NF1 и деформаций скелета в значительной степени неизвестна; однако сообщалось, что у пациентов с NF1 низкая минеральная плотность костей и низкая концентрация витамина D (2).
Исследования показали, что риск переломов у детей, страдающих NF1, увеличивается в три раза, а у взрослых-в пять раз (17, 18). Повторные переломы у этих пациентов могут привести к псевдоартрозу.
Желудочно-кишечные проявления. Желудочно-кишечный тракт может быть поражен нейрофибромами и злокачественными опухолями оболочек периферических нервов, аналогично другим участкам тела; однако также существуют различные другие злокачественные новообразования желудочно-кишечного тракта, связанные с нейрофиброматозом.
Что касается нейрофибром, желудочно-кишечный тракт может быть поражён очаговыми нейрофибромами или диффузной нейрофиброматозной пролиферацией, локализованной в собственной пластинке. В обоих случаях ганглиозные клетки могут пролиферировать без клинического значения.
Стромальные опухоли желудочно-кишечного тракта (GIST) не являются редким явлением у пациентов с NF1 и встречаются до 25% случаев (19). В отличие от GIST в общей популяции, GIST, ассоциированные с NF1, редко сочетаются с мутациями в KIT или PDGFRA.
В большинстве случаев GIST протекает бессимптомно и доброкачественно. Гистологически GIST имеет сходства с несиндромными опухолями, состоящие из веретенообразных клеток и скейноидных волокон.
Эндокринные опухоли желудочно-кишечного тракта также наблюдаются у пациентов с NF1, и они имеют склонность к локализации в периампулярной области.
Наиболее распространенной эндокринной опухолью, о которой сообщается, является соматостатинома; однако в этой ситуации также были описаны гастринома, инсулинома, карциноиды и параганглиомы (19).
Другие злокачественные новообразования. Пациенты с NF1 также имеют предрасположенность к злокачественным новообразованиям вне тканей нервной системы.
У детей с NF1 в семь раз повышен риск злокачественных новообразований кроветворной системы, особенно миелоидного лейкоза, по сравнению с их сверстниками. Лечение и прогноз аналогичны для общей популяции.
Пациенты с NF1 также подвержены повышенному риску развития рака молочной железы, особенно у женщин в возрасте до 50 лет.
Опять же, лечение одинаково для обеих групп пациентов. Наконец, хотя это редкое явление, феохромоцитомы надпочечников наблюдаются чаще у пациентов с NF1, чем в общей популяции, с зарегистрированной частотой до 5% по сравнению с менее чем 1% (2).
Проявление феохромоцитомы часто включает покраснение, учащенное сердцебиение и гипертонию, и хирургическое вмешательство часто является основным лечебным мероприятием.
Нейрофиброматоз 2 типа
Введение. NF2, также известный как двусторонний акустический нейрофиброматоз или центральный нейрофиброматоз, представляет собой наследственный опухолевый синдром, характеризующийся преимущественно развитием шванном, наряду с менингиомами, эпендимомами и аномалиями зрения.
Несмотря на название, нейрофибромы встречаются относительно редко.
NF2 наследуется по аутосомно-доминантному типу с предполагаемой частотой 1 на 25 000, распространенностью 1 на 60 000 и пенетрантностью приблизительно 0,95 (20).
Пациенты обычно находятся в возрасте около 20 лет, и прогностические аспекты включают возраст на момент постановки диагноза, фазу развития менингиомы и доступ к специализированным медицинским центрам (21).
Заболевание вызвано мутацией в гене NF2 на 22-й хромосоме, который кодирует белок мерлин. Более половины случаев вызваны мутациями гена de novo у пациентов без семейного анамнеза заболевания.
Диагностические критерии. Двусторонние шванномы верхней вестибулярной ветви восьмого черепного нерва (вестибулярная шваннома или акустическая неврома) являются патогномоничными для NF2.
Однако, поскольку у 41% пациентов, у которых в конечном итоге было доказано наличие NF2, на начальном этапе не было двусторонних вестибулярных шванном, было создано несколько диагностических стандартов для NF2.
К ним относятся широко признанные критерии Манчестера, а также дополнительные критерии NIH, приведенные в таблице 2. Бейзер и соавт. недавно предложили систему оценки для замены критериев Манчестера с якобы повышенной чувствительностью при сохранении 100% специфичности (21, 22).



