Синдром Шерешевского-Тернера
Данный синдром встречается с частотой 1:2000- 1:2500 новорожденных девочек.
На Х-хромосоме находятся гены, контролирующие развитие гонад, процессы роста и полового развития, нарушение их функции приводит к выраженной задержке роста, костным аномалиям и множественным порокам.
В зависимости от того, какой кариотип у пациентки, проявления синдрома могут варьировать от типичных признаков синдрома до их полного отсутствия.
Клинические признаки с. Шерешевского-Тернера:
1. Фенотипические особенности: птоз, эпикант, нистагм; высокое (готическое) небо; микрогнатия; деформация ушной раковины; короткая толстая шея; низкий рост волос на затылке, крыловидные складки шеи; «вдавленная» грудная клетка, широко расставленные, «втянутые» соски; сколиоз; отечность кистей/стоп (лимфостаз); пигментные невусы, гипертрихоз; витилиго; cubitus valgus; деформация Маделунга; вальгусная деформация ног; аллопеция; дисплазия ногтевых пластинок
2. Множественные пороки развития: пороки сосудов сердца (стеноз устья аорты, дилятация аорты, аневризма), пороки почек («подковообразная» почка, аплазия почки, аномалии сосудов почек).
4. Задержка полового развития (дисгенезия гонад, гипергонадотропный гипогонадизм). В результате хромосомных аномалий у девочек нарушается процесс созревания яичников, отмечается задержка или полное отсутствие полового созревания и, как следствие, развитие бесплодия. В некоторых случаях (чаще при кариотипе 45 Х/ 46 ХY) может развиваться гонадобластома.
5. Эндокринологические нарушения: аутоиммунный тиреоидит, гипотиреоз, нарушение толерантности к углеводам, гипергонадотропный гипогонадизм.
В редких случаях у пациентов с с.Шерешевского-Тернера встречается спонтанный пубертат.
Низкий рост пациентов с с.Шерешевского-Тернера не является результатом СТГ-дефицита и, в большинстве случаев, применение гормона роста с целью улучшить ростовой прогноз не приводит к выраженному улучшению динамики роста.
Диагностика:
1. Наличие фенотипических признаков синдрома.
2. Кариотип: 45 Х или другие варианты.
4. Врожденные пороки развития сердечно-сосудистой системы, почек
5. Дисгенезия гонад: по УЗИ недоразвитие матки и яичников; повышение уровня ЛГ, ФСГ крови; аменорея
Лечение:
2. Коррекция врожденных пороков развития.
3. При аутоиммунном тиреоидите в фазе гипотиреоза назначается L-тироксин.
4. При выявлении нарушений углеводного обмена назначается диета и при необходимости сахароснижающие препараты.
5. Назначение рекомбинантного гормона роста- вопрос дискутабельный.
Клинико-кариотипическая характеристика синдрома Шерешевского-Тернера
Полный текст:
Аннотация
Проанализировано 18 фенотипических признаков у 82 пациентов с синдромом Шерешевского-Тернера. Выделены минимальные диагностические признаки, встречающиеся с частотой не менее 805%. Сравнительный анализ распределения клинических признаков в группах с различными вариантами нарушений кариотипа показал, что группа пациентов с мозаичным кариотипом (45, X / 40, XX) была наименее нагружена патологическими признаками. Группы с кариотипом 48, Х и структурными нарушениями сходны по распределению фенотипических признаков. Был проведен многомерный статистический кластерный анализ матрицы суммарных фенотипических расстояний и выделен кластер, который позволил объединить 4 группы пациентов с различными нарушениями кариотипа. Результаты сопоставлены с опубликованными данными молекулярно-цитогенетических исследований.
Ключевые слова
Для цитирования:
Тарская Л.А., Прытков А.Н., Трубников В.И., Патютко Р.С., Касаткина Э.П., Козлова С.И. Клинико-кариотипическая характеристика синдрома Шерешевского-Тернера. Проблемы Эндокринологии. 1996;42(3):22-24. https://doi.org/10.14341/probl12038
For citation:
Tarskaya L.A., Prytkov A.N., Trubnikov V.I., Patyutko R.S., Kasatkina E.P., Kozlova S.I. Clinical and karyotypical characteristics of the Shereshevsky-Turner syndrome. Problems of Endocrinology. 1996;42(3):22-24. (In Russ.) https://doi.org/10.14341/probl12038
По данным исследователей разных стран, частота хромосомных аномалий у человека достаточно велика и составляет суммарно примерно 1 случай на 170 новорожденных [2]. Более половины этих аномалий связаны с нарушениями половых хромосом. Среди них значительный удельный вес составляет синдром Шерешевского — Тернера, который характеризуется выраженной вариабельностью клинических проявлений и разными вариантами хромосомных нарушений [1, 3, 10].
Такой клинический и цитогенетический полиморфизм затрудняет точную и своевременную диагностику синдрома Шерешевского — Тернера. Молекулярно-цитогенетические технологии, появившиеся в последние годы, расширили возможности для объяснения клинического полиморфизма этого синдрома благодаря созданию генетических карт половых хромосом [4, 7, 9].
В настоящем исследовании мы предприняли попытку проанализировать спектр клинических проявлений при синдроме Шерешевского — Тернера и сопоставить его с кариотипической вари- бельностью.
Материалы и методы
Обследовано 82 больных с синдромом Шерешевского — Тернера. Регистрация больных осуществлялась по обращаемости в научно-поликлиническое отделение Института клинической генетики МГНЦ РАМН с 1990 по 1994 г.
При обследовании на всех больных заполняли стандартную карту фенотипа по 15 признакам (табл. 1). Изучали в основном фенотипические признаки с альтернативным проявлением, количественные признаки для однородности сравнений были разделены на альтернативные группы, например нормальный и низкий рост, и т. п.
При проведении настоящего исследования применяли клинико-генеалогический метод, цитогенетические методы с диф
Распределение частоты фенотипических признаков у больных с синдромом Шерешевского—Тернера
Щитовидная грудная клетка
Низкий уровень роста волос
Гипоплазия грудных желез
Скудное вторичное оволосение
ференциальным окрашиванием (G-окраска). При статистической обработке данных были использованы стандартные статистические методы (средние и вариационные статистики), а также методы многомерного статистического анализа на основе пакета прикладных программ для ЭВМ, разработанного В. И. Трубниковым (1993 г.).
Результаты и их обсуждение
На первом этапе исследования была проанализирована частота встречаемости 15 фенотипических признаков среди всех больных с синдромом Шерешевского — Тернера (см. табл. 1).
Как видно из данных, представленных в табл. 1, основными признаками (встречающимися с частотой более 50%) являются низкий рост, щитовидная грудная клетка, первичная аменорея, гипоплазия грудных желез и скудное вторичное оволосение.
Из сопутствующих соматических аномалий наиболее часто встречались короткая шея, шейный птеригиум, низкий уровень роста волос на шее, высокое небо, брадидактилия, гипоплазия ногтей, cibitus valgus, частота встречаемости которых варьирует от 20 до 45%. Всего среди 82 больных с синдромом Шерешевского — Тернера встретилось 15 цитогенетических вариантов нарушений кариотипа. Причем основной удельный вес (80%) составляли различные структурные аномалии.
Как видно из приведенных данных, моносомия 45,X составляет 29,27% среди всех больных с синдромом Шерешевского — Тернера.
Анализ распределения 15 фенотипических признаков в группе больных с синдромом Шерешевского — Тернера представлен в табл. 2.
Статистически достоверные различия между группами были обнаружены по 9 признакам фенотипа из 15 изученных.
Низкий рост был характерен для большинства больных 1-й (95,8%) и 3-й (100%) групп и реже встречался во 2-й группе (62,5%).
Распределение частоты фенотипических признаков при разных вариантах кариотипа у больных с синдромом Шерешевского—Тернера
Щитовидная грудная клетка
Низкий уровень роста волос
Гипоплазия грудных желез
Скудное вторичное оволосение
Укорочение шеи также реже отмечалось в группе больных с мозаичным кариотипом, сопровождающимся количественными перестройками » (25%), и наиболее часто проявлялось при «чистой»
Шейный птеригиум приблизительно с одинаковой частотой встречался во 2-й и 3-й группах (12,5 и 10% соответственно) и примерно в 4 раза чаще — в 1-й группе (45,8%).
Эпикант практически не отмечен во 2-й группе, с небольшой частотой (8%) встречался в 3-й группе и отмечался у 1/3 (33,3%) больных 1-й группы.
Гипоплазия ногтей чаще встречалась в 1-й группе (50%), несколько реже — в 3-й (38%) и не наблюдалась во 2-й группе.
Первичная аменорея также чаще наблюдалась в 1-й (92,3%) и в 3-й (87,1%) группе по сравне- ( нию со 2-й (50%).
Аналогичным образом распределялись по группам такие признаки, как гипоплазия грудных желез и скудное вторичное оволосение. Эти признаки чаще встречались в 1-й и 3-й группах по сравнению со 2-й.
Таким образом, суммарный анализ данных по всем признакам позволяет сделать вывод о том, что наименее нагруженной патологическими признаками является 2-я группа (мозаицизм с наличием нормального клона 46,XX). Очевидно, наличие нормального клона клеток частично компенсирует клинические проявления большинства изучаемых признаков синдрома.
Можно отметить, что 3-я группа по общей оценке нагруженности признаками занимает как бы промежуточное положение между 1-й и 2-й, но в то же время характеризуется большей вариабельностью по сравнению с ними.
При попарном сравнении частот встречаемости между группами наиболее существенные и часто встречающиеся различия затрагивали гипоплазию ногтей. Этот признак достоверно чаще встречался в 5-й группе по сравнению с 1-й (р <0,05); в 3-й и 4-й группах (р <0,01). По-видимому, наличие idic(X) связано с нарушениями развития тканей мезодермального происхождения, костей, соединительной ткани и соматических клеток гонад. По данным [3], частота этого признака (гипоплазия ногтей) также оказалась выше у больных с указанным кариотипом, чем с моносомией X.
Кроме того, обнаружены тенденция на границе статистической достоверности к большей частоте cubitus valgus в 1-й группе по сравнению с 3-й и отставание в росте в 1-й группе по сравнению с 4-й. Последнее, возможно, связано с утратой Xq.
Связи остальных групп выражены слабее. Их кариотипы на дендрограмме соответствуют: 5 —
О.’ПОЗ О.235Г О
Кластерный анализ матрицы обобщенных расстояний между группами больных с разными вариантами кариотипа.
Наиболее фенотипически близкими оказались варианты кариотипов с утратой части материалов Х-хромосомы (разные варианты делений).
В литературе имеются предположительные данные о локализации некоторых локусов, детерминирующих тернеровский фенотип в области Хсеп- pil, обозначенный как критический регион по данному синдрому [8]. Также на Y-хромосоме обнаружен критический регион, ассоциирующийся с некоторыми признаками синдрома Шерешевского — Тернера (cubitus valgus, щитовидная грудная клетка, короткие 4 метакарпальные кости), локализованный на Yp subintervals 5А—51 [5].
Прогресс в совместном использовании классических цитогенетических и современных молекулярно-генетических методов позволяет надеяться на выявление роли отдельных регионов половых хромосом в объяснении широкого полиморфизма синдрома Шерешевского — Тернера и происхождении отдельных клинических симптомов.
3-я группа больных (структурные аномалии Х-хромосомы) по общей оценке нагруженности г признаками занимает как бы промежуточное положение между 1-й (моносомия 45,X) и 2-й группами, но в то же время характеризуется большей вариабельностью по сравнению с ними.
Фертильность у женщин с синдромом Шерешевского-Тернера.
Кариотип – полный набор хромосом диплоидной клетки (кариотип человека в норме 46,ХХ или 46,ХY).
Фенотип – совокупность признаков организма, контролируемый определенным генотипом.
Стигмы – фенотипические отклонения от нормального развития.
Фертильность – детородная функция живого организма.
Гоносомы – половые хромосомы это Х- и Y-хромосома.
Моносомия – утрата целой хромосомы.
Стреки – соединительнотканные тяжи.
Центромера – органоид, располагающийся в районе первичной перетяжки хромосомы. Она делит хромосому на два плеча.
Теломера – концевой участок хромосомы. Специализированная структура, обеспечивающая стабильность линейной молекулы ДНК.
Делеция – утрата участка хромосомы.
Дисгенезия –нарушение формирования половой железы и вторичных признаков пола.
Полная или частичная потеря одной хромосомы Х ведет большинстве случаев к абсолютному бесплодию. Но в 1960 г. F.Bahner и соавт. [2] впервые сообщили о фертильности у пациентки с синдромом Шерешевского–Тернера и с кариотипом 45,Х0. С этого времени описано более 100 беременностей у женщин с синдромом Шерешевского–Тернера. При этом беременные имели в большинстве гоносомальный мозаицизм между одной клеточной линией с различной аберрантной хромосомой Х или моносомией Х и одной нормальной линией клеток 46,ХХ, причем отношения обеих клеточных линий были различными. Дети матерей с мозаицизмом, например 46,ХХ/45,Х, имели как нормальный кариотип [13], так и аберрантный гоносомальный клон клеток [3]. Эти данные говорят, что потеря одной полной хромосомы Х или какой-то одной части хромосомы Х, не в каждом случае должна сопровождаться бесплодием [3, 14].
Приводим наши наблюдения фертильности у женщин с бесплодием, невынашиванием, имеющих детей с врожденными пороками развития в анамнезе, с мозаичным кариотипом 46,ХХ/45,Х.
Больная К. обратилась в медико-генетический центр 26.07.95 с целью генетического обследования и решения вопроса о прогнозе будущего потомства.
Из акушерского анамнеза выяснено, что 29.08.95 в сроке 26–27 нед была прервана беременность. Причиной для прерывания беременности послужило заключение ультразвукового исследования (УЗИ), которое выявило множественные врожденные пороки развития плода: укорочение нижних конечностей, двухкамерное сердце, гидроцефалия. После прерывания беременности у плода мужского пола, массой 0,925 кг, были установлены все выявленные на УЗИ пороки развития.
Из анамнеза женщины: менархе в 12 лет, месячные спонтанные, регулярные, через 24 дня по 3 дня, безболезненные, умеренные. Половая жизнь с 21 года. Беременность наступила спонтанно в течение первого года регулярной половой жизни и закончилась 29.08.95 в сроке 26–27 нед прерыванием беременности по медицинским показаниям.
Генеалогический анамнез семьи пробанда представлен на рис. 1.
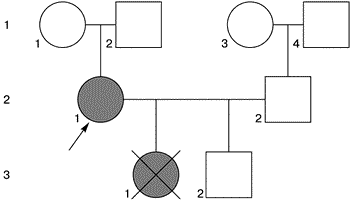 |
| Рис 1. Схема родословной семьи пробанада К |
Данные обследования. Женщина правильного телосложения, рост 160 см, масса 56 кг. Вторичные половые признаки развиты удовлетворительно. Отмечается неправильный прикус, неправильный рост зубов нижней и верхней челюсти, высокое небо. Значительных стигмальных отклонений не выявлено.
При хромосомном анализе у мужа нормальный мужской кариотип 46,XY. В кариотипе пробанда выявлена патологический клон клеток с моносомной X-конституцией – 11% (кариотип 46,XX/45,X – 11%).
23.06.97 больная вновь обратилась в медико-генетический центр по поводу прогноза потомства в сроке беременности 30 нед. При УЗИ патологии со стороны внутриутробного плода не выявлено. Гормональный скрининг и инвазивная пренатальная диагностика не проводились в связи с обращением в центр в позднем сроке беременности. Беременность пролонгировалась под наблюдением врача женской консультации.
14.10.97 срочные роды без осложнений в срок 39 нед беременности. Родился здоровый мальчик массой 4450 г и длиной тела 54 см. Из родильного дома был выписан домой на 8-е сутки жизни. При осмотре ребенка в медико-генетическом центре патологических отклонений в фенотипе не выявлено. Кариотипический анализ лимфоцитов периферической крови ребенка показал нормальный мужской хромосомный набор 46,XY, без числовых и структурных перестроек.
Больная Б. 22 года, медицинская сестра. Обратилась в медико-генетический центр 5.09.95 с жалобами на отсутствие беременности в течение 1 года после замужества. Состоит в первом неродственном браке. Мужу 22 года.
Из анамнеза выяснено: менархе в 12 лет, месячные нерегулярные: интервал от 32 до 44 дней, менструации по 4 дня, очень болезненные. Половая жизнь с 21 года без контрацепции.
Генеалогический анамнез представлен на рис. 2.
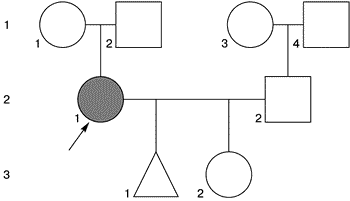 |
| Рис 2. Схема родословной семьи пробанада Б |
Данные обследования. Правильного телосложения, рост 154 см, масса 56 кг. При осмотре: диспластичные ушные раковины, выступающий фильтр, диастема, высокое небо, низкий рост волос на шее, широкий плечевой пояс, дисплазия ногтевых пластин.
Половая формула Ме+ Ма2 Ах3 Рв3.
УЗИ внутренних гениталий (на 11-й день цикла): матка размерами 47′ 30′ 38 мм, грушевидной формы, шейка матки 25 мм, эндометрий 7 мм, правый яичник 47′ 25′ 25 мм, левый – 46′ 26′ 26 мм, содержит множество жидкостных включений в диаметре до 5–6 мм.
Гормоны крови (7-й день цикла): тестостерон 11,3 нм/л, пролактин 446 нг/мл, ЛГ 19,7 мМЕ/мл, ФСГ 5,5 мМЕ/мл, ТТГ 2,8 МЕ/л.
Кариотип – 46,ХХ/45,Х – 15%, кариотип мужа 46,XY, без числовых и структурных перестроек.
Через 7 мес после первого обращения в медико-генетический центр спонтанно наступила беременность, которая самопроизвольно прервалась в сроке 5–6 нед.
Через 2 года после индукции овуляции клостильбегитом в дозе 50 мг в день с 5-го по 9-й день цикла наступила беременность. При медико-генетическом консультировании рекомендовано проведение комплекса мероприятий по пренатальной диагностике.
При гормональном исследовании крови по программе пренатальной диагностики «АЛЬФА» в срок беременности 16–17 нед от 8.02.99: концентрация a-фетопротеина – 37,6 МЕ/мл – 0,95 МОМ; ХГ – 47 340 МЕ/л – 1,64 МОМ; Е3 – 33,6 нмоль/л – 1,05 МОМ.
Результат скрининга: риск рождения ребенка с болезнью Дауна 1:1 300; риск рождения ребенка с дефектом невральной трубки – 1:12 000.
При УЗИ плода в срок беременности 23 и 34 нед патологии не выявлено.
С целью исключения хромосомной патологии плода 19.01.99 беременной был произведен трансабдоминальный кордоцентез. Анализ кариотипирования лимфоцитов крови плода показал нормальный женский кариотип 46,ХХ, без числовых и структурных перестроек. Учитывая наличие нормального кариотипа у плода, женщина пролонгировала беременность.
Роды произошли в срок беременности 38 нед. Родилась здоровая девочка массой 3300 г, длиной 54 см.
По современным представлениям, гонады эмбриона с кариотипом 45,Х дегенерируют в позднем фетальном периоде или в раннем периоде новорожденности и представляют собой типичные гонады тяжи [1]. Период до полной атрезии всех фолликулов может длиться до 20 или 30 лет, поэтому существование активных фолликулов при дисгенезии гонад возможно и у взрослых индивидуумов. Клиническая картина зависит от структуры дисгенетичной гонады. Возможен спонтанный пубертат как в представленных случаях и/или ранняя вторичная аменорея. Так, A.M. Pasquino и соавт. сообщают о 522 девочках с синдромом Шерешевского–Тернера, старше 12 лет, не имеющих отчетливых признаков заболевания, с кариотипом 45,Х и 45,X/46,XX [12]. По данным этих авторов, у 84 больных был спонтанный пубертат с менархе в 13 лет. При этом костный возраст в среднем составил 12,9 года. Регулярный менструальный цикл сохранялся у 30 больных в среднем 9,2 года после менархе. Вторичная аменорея развилась в среднем через 1,6 года. У некоторых менструации приобрели нерегулярный характер через 9 мес. У 3 больных наступила беременность, которая закончилась в одном случае рождением здорового ребенка, в другом – рождением ребенка с синдромом Тернера и в третьем – рождением ребенка с врожденными пороками развития.
C.Huseman [7] сообщает даже о случае с преждевременным пубертатом, с менархе в 10-месячном возрасте при кариотипе 45,Х/46,ХХ. Возможно имело место персистирование функционально полноценного фолликула при наличии клеточной линии с нормальным кариотипом.
Отметим, что в представленных случаях у больных был мозаичный кариотип с преобладанием нормальной ХХ-клеточной линии в лимфоцитах периферической крови.
Нормальное развитие яичников во время фетального периода с наступающей после рождения регрессией описано у девочек с хромосомной конституцией 46,X,i (Xq) [6].
A.Farabosco и соавт. указывают, что хромосомные участки Xq13- больше q21 и Хq21- больше q26 ответственны за развитие фертильных гонад. Авторы приводят данные о фертильности женщин с гетерозиготным дефицитом сегмета Хр ter- больше p21 [5]. Возможно, гены, необходимые для поддержания функциональной способности ооцитов, локализованы между Хp21 и центромерой [6].
Около 5–11% всех фертильных женщин с синдромом Шерешевского–Тернера имеют кольцевую хромосому Х с кариотипическим мозаицизмом 45Х/46,Х,r(X). Происхождение радиарной хромосомы обусловлено делецией обеих теломер или теломерных сегментов с присоединенным слиянием мест переломов хромосом X или Y [8].
E.Bannier и соавт. [3] сообщают о случае передачи кольцевой хромосомы Х от фергильной матери к дочери. Современные методы определения происхождения подобной хромосомы (неауторадиографическая гибридизация in situ биотинированных Х (qSV2X5)- и Y (Y84)-специфических для центромер зондов) позволяют четко и быстро идентифицировать кольцо Х-происхождения [4] у матери и потомка.
Описан также случай синдрома Шерешевского–Тернера у пробанда с кариотипом 45,Х/46,Х, del (X) (q21.2-q ter)/46,X, r(X), имеющего функциональную дисомию и изодисомию материнского происхождения [9], что указывает на возможность передачи аберрантной хромосомы от фертильнои матери к потомству.
Ребенок от женщины с мозаичным вариантом синдрома Шерешевского–Тернера имел в нашем случае нормальный кариотип. Это позволяет считать, что созревшая яйцеклетка должна содержать неаберрантную хромосому Х. Эту мысль подтверждает A.Novac и соавт. [11] и A.Mori и соавт. [10], которые нашли разную патологию в хромосомном статусе периферической крови, кожных фибробластов, а также левой и правой гонад. Таким образом, кариотип, определенный в лимфоцитах периферической крови, не в каждом случае определяет ситуацию в гонадах.
Наличие у женщин кариотипа с патологическим клоном клеток (с мозаичным или моносомным вариантом синдрома Шерешевского–Тернера) не исключает наступления у них спонтанных, неиндуцированных беременностей и рождения здоровых детей. Разнообразие в экспрессии клинической картину синдрома Шерешевского–Тернера удивительно. Оно не характеризуется только тернеровскими стигмами и степенью гонадальной дисгенезии. При синдроме Шерешевского–Тернера спонтанные менструации при патологической гоносомной конституции встречаются чаще, чем нормальная фертильность.
Т.А. Соколова, Г.Г. Шупта, М.П. Корф
Красноярский межрегиональный медико-генетический центр
Проблемы долгосрочного лечения больных синдромом Шерешевского–Тернера
Синдром Шерешевского–Тернера (СШТ) — генетически детерминированное заболевание, развивающееся в результате качественного либо количественного нарушения одной половой Х-хромосомы.
 Синдром Шерешевского–Тернера (СШТ) — генетически детерминированное заболевание, развивающееся в результате качественного либо количественного нарушения одной половой Х-хромосомы. По данным разных авторов частота встречаемости заболевания колеблется от 1:2000 до 1:2500 новорожденных девочек. Классическое описание СШТ было сделано американским врачом Генри Тернером в 1938 году. Однако еще раньше, 12 ноября 1925 года, профессор Н. А. Шерешевский представил клинические данные женщины 25 лет: низкий рост (132 см), выраженный половой инфантилизм — первичная аменорея, отсутствие вторичных половых признаков, недоразвитие внутренних гениталий.
Синдром Шерешевского–Тернера (СШТ) — генетически детерминированное заболевание, развивающееся в результате качественного либо количественного нарушения одной половой Х-хромосомы. По данным разных авторов частота встречаемости заболевания колеблется от 1:2000 до 1:2500 новорожденных девочек. Классическое описание СШТ было сделано американским врачом Генри Тернером в 1938 году. Однако еще раньше, 12 ноября 1925 года, профессор Н. А. Шерешевский представил клинические данные женщины 25 лет: низкий рост (132 см), выраженный половой инфантилизм — первичная аменорея, отсутствие вторичных половых признаков, недоразвитие внутренних гениталий.
Основными задачами лечения больных с СШТ в детском и подростковом возрасте являются увеличение конечного роста, формирование вторичных половых признаков с установлением регулярного менструального цикла, коррекция пороков развития и профилактика остеопороза. Своевременно начатая и адекватно проводимая терапия обеспечивает в последующем женщинам с СШТ полноценную активную жизнь. Однако многообразие клинических проявлений заболевания, неоднородность патофизиологических механизмов их развития создают проблемы в достижении оптимальных результатов лечения.
Наряду с низкорослостью и гипергонадотропным гипогонадизмом у больных с СШТ часто наблюдаются пороки развития внутренних органов, обусловленные хромосомными аберрациями. По мнению некоторых авторов, хромосомные аномалии, характерные для СШТ, могут иметь до 3% всех эмбрионов женского пола, однако вследствие спонтанных абортов лишь 1% плодов сохраняется до конца беременности. Таким образом, СШТ ответственен за 7–10% всех самопроизвольных прерываний беременности [1].
Гипогонадизм встречается почти у 98% больных СШТ. Редко диагностируют сохранную функцию яичников, проявляющуюся, как правило, увеличением молочных желез. Для первичной овариальной недостаточности, развивающейся в результате дисгенезии гонад, характерно значительное увеличение уровня гонадотропных гормонов в результате отсутствия отрицательного подавляющего влияния половых гормонов на гипоталамо-гипофизарную систему. У девочек с СШТ уже в первые недели жизни определяется повышение уровня гонадотропных гормонов, преимущественно фолликулостимулирующего гормона (ФСГ). Такое повышение сохраняется до двухлетнего возраста. В возрасте от 2 до 6 лет происходит снижение уровня гонадотропных гормонов в результате активации универсального центрального механизма, подавляющего секрецию гонадотропин-релизинг-гормона (ЛГ-РГ) независимо от уровня половых гормонов. В возрасте 5–6 лет начинают вновь повышаться ФСГ и лютеинизирующий гормон (ЛГ). В период полового развития отмечено увеличение уровня гонадотропных гормонов (ФСГ, ЛГ) в 10 раз [1, 3, 4, 14].
У 5–7% девочек с гипергонадотропным гипогонадизмом может отмечаться спонтанное половое развитие, чаще при мозаичном варианте хромосомного набора. У этих девочек спонтанное половое развитие чаще является неполным и не приводит к нормальной и длительной овариальной функции. Однако в литературе описаны случаи спонтанных и повторных беременностей и родов у женщин с СШТ. Необходимо отметить, что проявлением истинного пубертата следует считать только увеличение молочных желез. Вторичное оволосение — лобковое и аксиллярное — развивается спонтанно у всех девочек с гипергонадотропным гипогонадизмом к 12–13 годам под влиянием надпочечниковых андрогенов [1, 3].
Больные с СШТ в 100% случаев имеют низкий рост. Девочки уже при рождении имеют низкую массу и длину тела. В течение первых 2–3 лет скорость роста достаточно стабильна. В последующем в возрасте от 3 до 11 лет отмечается выраженное снижение темпов роста. В возрасте пубертата ростового скачка не происходит, и отставание в росте становится максимальным [1, 14, 22, 23]. До недавнего времени патогенез низкорослости при СШТ оставался неясным.
Существовал ряд гипотез, объясняющих этот симптом заболевания. При этом влияние различных вариантов кариотипа при СШТ на конечный рост доказано не было. В последнее десятилетие было установлено, что ведущую роль в патогенезе низкорослости у пациентов с гипергонадотропным гипогонадизмом имеют непосредственные генетические нарушения, обусловленные делецией гена SHOX. Это ген, локализованный в псевдоаутосомной области на конце короткого плеча Х-хромосомы, с которым связывают низкорослость при СШТ и некоторых других синдромах, сопровождающихся мезомиелической дисплазией костной ткани. Ген SHOX (short stature homeobox gene) кодирует белок, содержащий гомеодомен, который с наибольшей вероятностью работает как регулятор транскрипции. Снижение экспрессии SHOX-гена может быть ассоциировано не только с низкорослостью, но и с деформацией скелета (деформация Маделунга, вальгусная деформация, микрогнатия, «готическое небо», укорочение конечностей и пястных костей) [3].
Пациенты с СШТ требуют не только коррекции гонадной недостаточности, но и стимуляции роста. Выраженная задержка роста начинает формироваться у детей с 5–6 лет, и конечный рост колеблется около 140 см.
Соматотропная функция у больных с СШТ на фоне стимуляционных проб продемонстрировала адекватный выброс соматотропного гормона (СТГ) в ответ на стимуляцию. У девочек с СШТ снижение секреции СТГ до периода полового развития происходит в результате дефицита эстрогенов. У пациентов также было обнаружено снижение уровня инсулиноподобного фактора роста-1 (ИФР-1), необходимого для осуществления периферического действия соматотропного гормона [1, 2, 5, 6]. Некоторые ученые полагают, что у больных с гипергонадотропным гипогонадизмом отмечается частичная резистентность к гормону роста и ИФР-1. Однако это требует дальнейшего изучения.
В мировой практике в настоящее время начато широкое применение рекомбинантных препаратов гормона роста у девочек с СШТ. Многочисленные исследования показали, что препараты соматропина ускоряют рост у этих детей и увеличивают их конечное значение до 150–155 см. Увеличение конечного роста в результате лечения супрафизиологическими дозами соматропина по данным разных авторов весьма вариабельно — от 3 до 16 см по отношению к прогнозируемому без лечения росту [14, 22, 25, 26]. В связи с тем, что спонтанный пубертат развивается лишь у 20% девочек с СШТ, возникает необходимость индукции полового развития с помощью эстрогенов. При этом эстрогены активируют закрытие эпифизарных зон роста и являются лимитирующим фактором роста костей в длину. Исследования ряда авторов свидетельствуют о том, что присоединение к терапии гормоном роста препаратов эстрогенов не оказывает влияния на показатели конечного роста пациентов.
Так, по данным Rosenfeld R. и соавт. низкие дозы парентерально введенного депо-эстрадиола на фоне лечения гормоном роста способствуют феминизации пациенток без влияния на конечный рост [21]. Вместе с тем имеются доказательства того, что раннее назначение эстрогенов на фоне лечения гормоном роста приводит к уменьшению величины конечного роста девочек с СШТ. Следовательно, назначение половых стероидов должно быть отложено насколько это возможно, несмотря на то, что отсроченное назначение эстрогенов может привести к нежелательным последствиям и замедлению костной минерализации [15]. В связи с этим до настоящего времени вопрос о сроках начала терапии гормоном роста и препаратами эстрогенов окончательно не решен. Основные рекомендации по вопросам лечения гормоном роста сводятся к тому, что необходимо начинать терапию соматропином, как только рост пациента с СШТ окажется ниже 50-й перцентили кривой роста здоровых девочек и может начинаться даже с 2-летнего возраста. Заместительная гормональная терапия эстрогенами должна начинаться в возрасте не ранее 12 и не позднее 15 лет [26].
Belgian Study Group for Pediatric Endocrinology провела ретроспективное исследование показателей роста и полового развития у 186 больных с СШТ в зависимости от сроков начала терапии гормоном роста и препаратами эстрогенов. Результаты свидетельствовали о том, что лечение гормоном роста приводило к увеличению окончательного роста у большинства девочек с СШТ. Окончательный рост у леченных пациентов составил в среднем 151,7 ± 6,0 см, что на 8,3 см выше, чем у нелеченных девочек с СШТ. При этом возраст начала терапии гормоном роста не влиял на конечный рост. Также не оказывал влияния на показатели конечного роста спонтанный или отсроченно индуцированный эстрогенами пубертат [19].
В соответствии с принятыми на V Международном совещании по синдрому Тернера в 2000 г. рекомендациями, наиболее эффективным средством лечения низкорослости у больных с гипергонадотропным гипогонадизмом являются препараты гормона роста человека (Генотропин, Нордитропин, Хуматроп, Сайзен), которые вводятся ежедневно подкожно в вечерние часы в суточной дозе 0,05 мг/кг массы тела. Терапию гормоном роста прекращают, когда костный возраст достигает 15 лет, а скорость роста падает до 2 см в год.
Заместительную терапию эстрогенами рекомендуют начинать в возрасте 14–15 лет, когда достигнут рост, близкий к конечному. Препаратами выбора для инициации пубертата у девочек в настоящее время являются конъюгированные эстрогены (Премарин 625 мкг в сутки) и производные бета-эстрадиола (Эстрофем, Прогинова 1 мг в сутки). Возможно также наружное применение эстрогеновых препаратов в виде гелей (Дивигель). Через 12–18 месяцев монотерапии эстрогенами переходят к циклической терапии эстроген-прогестагеновыми препаратами [1, 3, 9, 25]. Длительность заместительной гормональной терапии — до возраста наступления менопаузы у здоровых женщин (до 50 лет) [10]. Адекватность индивидуального подбора лечения оценивают через 3 и 6 месяцев.
Широкое развитие методов экстракорпорального оплодотворения позволяет женщине с СШТ вынашивать беременность и рожать детей. При достижении адекватного полового развития возможно возникновение беременности путем экстракорпорального оплодотворения с помощью донорской яйцеклетки. Для девочек, имеющих спонтанный, но кратковременный период овариальной активности, возможна криоконсервация ооцитов с последующим их оплодотворением и внедрением в эндометрий пациентки, желающей иметь ребенка. В подобных случаях необходимо предварительное генетическое тестирование эмбриона.
У девочек старше 8 лет лечение гормоном роста рекомендуют сочетать с оксандролоном — неароматизирующимся анаболическим стероидом, обладающим минимальной андрогенной активностью [24]. К настоящему времени в литературе отсутствуют результаты долгосрочных наблюдений по применению гормональной заместительной терапии препаратами эстрогенов и лечению гормоном роста в сочетании с оксандролоном при СШТ. Вместе с тем известен большой ряд проблем, возникающих в течение жизни этих больных, связанных с клиническими особенностями заболевания.
При СШТ общая заболеваемость пациенток выше, чем в популяции. Относительный риск (ОР) развития эндокринных заболеваний выше в 4,9 раза. В том числе риск развития аутоиммунного тиреоидита превышает 16 раз, сахарного диабета 1-го типа — 11 раз, сахарного диабета 2-го типа — 4 раза. Более чем в два раза у этих пациенток увеличен риск развития ишемической болезни сердца и гипертонии, сосудистых заболеваний мозга и атеросклероза. Наблюдается повышенный риск развития цирроза печени (ОР — 5,7), остеопороза (ОР — 10,1). Относительный риск возникновения всех раковых заболеваний составляет 1,35, в то время как риск развития рака толстой и прямой кишки значительно повышен (4,94). По итогам когортного исследования, проведенного в Великобритании, относительный риск смерти у больных с СШТ превысил 4 раза вследствие заболеваний нервной, пищеварительной, сердечно-сосудистой, дыхательной и мочеполовой систем [27].
Большую долю в увеличении заболеваемости и смертности при СШТ отводят врожденным и приобретенным заболеваниям сердца. Характерными для СШТ являются такие врожденные пороки, как коарктация аорты, двустворчатый аортальный клапан. Двустворчатый аортальный клапан является наиболее частой находкой и встречается у 13–34% больных с СШТ, в общей популяции этот порок встречается с частотой 1–2%. Коарктацию аорты обнаруживают у 4–14% больных с СШТ, преимущественно при кариотипе 45Х. По данным V. P. Sybert у 56% больных с СШТ выявляется патология сердечно-сосудистой системы. Врожденные пороки сердца, такие как: коарктация аорты, незаращение межжелудочковой и межпредсердной перегородок, транспозиция сосудов, сердца, могут приводить к гемодинамическим нарушениям [3, 14, 16, 22].
У 30% девочек с СШТ при амбулаторном суточном мониторировании артериального давления выявляется артериальная гипертензия легкой степени, у 50% выявляется ненормальный профиль суточных колебаний артериального давления. У взрослых пациенток с СШТ было отмечено значительное повышение артериального давления по сравнению со сходной по возрасту контрольной группой, причем у половины из них была клинически выраженная артериальная гипертензия [20]. Несмотря на то, что ишемическая болезнь сердца и цереброваскулярные нарушения у больных с СШТ встречаются значительно чаще, чем в популяции, показатели липидного спектра крови при этом синдроме не отличаются от таковых в группе контроля.
В литературе описано более 60 случаев дилятации или расслоения аорты [11]. Увеличение заболеваемости сердечно-сосудистыми заболеваниями связывают с хронической недостаточностью эстрогенов. Вместе с тем назначение гормональной заместительной терапии препаратами эстрогенов и лечение гормоном роста определяют необходимость углубленного кардиологического обследования с учетом возможного влияния терапии на функциональное состояние сердца и магистральных сосудов. Принимая во внимание развитие программ экстракорпорального оплодотворения, можно предположить увеличение в будущем количества беременностей у пациенток с СШТ [12]. При этом беременность может увеличить риск последующего расслоения аорты.
У 80% пациенток среднего возраста с СШТ наблюдается повышение уровня печеночных ферментов в крови, которое не связывают с заболеваниями печени. Вместе с тем результаты эпидемиологических исследований Gravholt C. H. et al. свидетельствуют о повышенной частоте цирроза печени при СШТ. При изучении биоптатов печени у 27 больных с СШТ, имевших повышенные уровни печеночных ферментов, были обнаружены различные патологические морфологические изменения, включая выраженную узловую регенеративную гиперплазию, множественную фокальную узловую гиперплазию, цирроз с облитерирующей портальной флебопатией, портальный фиброз, воспалительные инфильтраты, неалкогольный жировой гепатоз.
Авторы сделали заключение о том, что основной причиной таких морфологических изменений в печени при СШТ являются сосудистые нарушения врожденного характера и неалкогольный жировой гепатоз. При этом была исключена роль терапии эстрогенами в формировании патологии. Более того, лечение половыми стероидами независимо от пути их введения (таблетированная или трансдермальная формы 17 бета-эстрадиола) приводило к снижению повышеннных уровней сывороточной аланинаминотрансферазы, гамма-глутаминтрансферазы, щелочной фосфатазы. Показатели снижались в большей степени при терапии этинилэстрадиолом по сравнению с конъюгированными эстрогенами. Полученные результаты позволили сделать заключение о протективном действии эстрогенов в поддержании нормальной функции печени у пациенток с СШТ [13].
Большое количество исследований посвящено вопросам состояния костной системы и минерального обмена при СШТ. Исследователи обнаружили, что остеопороз и переломы при этом заболевании встречаются значительно чаще, чем в общей популяции. При этом снижение минеральной плотности костей повышает риск возникновения переломов любой локализации и в любом возрасте, включая детский. Механизм развития остеопороза при данной патологии до конца не установлен. Гормональные нарушения, наблюдаемые при СШТ, способствуют формированию остеопороза [4, 8]. Дефицит эстрогенов в период полового развития подавляет эндоостальный рост кортикального слоя костной ткани. Это приводит к нарушению формирования трабекулярной зоны кости. При сохраняющемся низком уровне эстрогенов у больных система соматотропный гормон — инсулиноподобный фактор роста в период полового развития не активизируется. В результате дефицита гормона роста и ИФР-1 происходит недостаточное образование костной ткани.
Рядом работ показано, что адекватная заместительная терапия препаратами эстрогенов в сочетании с гормоном роста способствует нормализации костной массы у больных с СШТ [25]. Вместе с тем до настоящего времени не опубликовано результатов долгосрочных исследований по изучению эффектов эстрадиола в сочетании с гормоном роста при лечении СШТ. Необходимость таких исследований определяется формированием оптимальной терапии в подростковом возрасте для достижения двух целей. Первая заключается в нормализации костной массы и сохранении минеральной плотности костей без риска влияния на конечный рост.
Вторая — своевременная индукция полового созревания для формирования вторичных половых признаков. Исследования Khastgir et al. показали, что применение 2 мг эстрадиола или его эквивалента для заместительной гормональной терапии при СШТ является недостаточным как в отношении формирования оптимальной костной массы, так и в отношении формирования вторичных половых признаков. Однако неизвестно, как более высокие дозы эстрогенов могут влиять на другие показатели у взрослых пациенток [18].
У значительной части подростков и взрослых больных с СШТ выявляются различные нарушения углеводного обмена. Чаще встречаются нарушенная толерантность к глюкозе и сахарный диабет 2-го типа [7, 17]. Согласно современным рекомендациям гормональная заместительная терапия препаратами эстрогенов при СШТ продолжается до возраста естественной менопаузы (50–55 лет). При этом сведений о долгосрочном влиянии эстрогенов на показатели углеводного обмена нет.
Очевидно, что основными задачами лечения этих пациентов являются увеличение конечного роста, формирование вторичных половых признаков, профилактика остеопороза. Однако остается открытым вопрос о возрасте, когда необходимо начинать лечение, о дозах и схемах комбинации препаратов, стимулирующих рост и половое созревание и предотвращающих развитие осложнений со стороны костной системы.



