Муковисцидоз: что за болезнь, как проходит лечение?
Симптомы муковисцидоза
Специалисты выделяют несколько форм заболевания: кишечную, легочную и смешанную.
К основным симптомам заболевания обычно относят:
Симптомы муковисцидоза зависят от формы болезни. Муковисцидоз легких сильно снижает иммунитет, а поскольку слизистые пробки легко поражаются бактериями стафилококка или синегнойной палочки, люди постоянно страдают от повторяющихся бронхитов и пневмоний.
Основные признаки муковисцидоза легких 1 :
Кишечная форма муковисцидоза характеризуется ферментной недостаточностью, нарушением всасывания полезных веществ в кишечнике. В результате нехватки ферментов стул становится «жирным», могут активно размножаться бактерии, в результате чего накапливаются газы, что приводит к выраженному метеоризму. Стул учащается, а объем каловых масс может в несколько раз превышать возрастную норму.
Причины муковисцидоза

Скопление слизи создает благоприятные условия для размножения условно-патогенной флоры, поэтому резко возрастает риск возникновения гнойных осложнений и сопутствующих заболеваний. При муковисцидозе у взрослых значительно страдает репродуктивная функция.
Диагностика муковисцидоза
Для диагностики муковисцидоза могут понадобиться как лабораторные, так и генетические обследования. Генетические исследования обладают высокой информативностью и позволяют заподозрить развитие болезни почти сразу после рождения. На данный момент обнаружить заболевание можно еще до рождения ребенка при неонатальном скрининге.
Для того, чтобы с уверенностью говорить о наличии у человека муковисцидоза, врач должен диагностировать следующие показатели 1 :
Лечение муковисцидоза
Лечение муковисцидоза должно быть комплексным и направленным на основные симптомы болезни: борьбу с инфекционными заболеваниями, очищение бронхов от мокроты, восполнение недостающих ферментов поджелудочной железы.
Часть терапии обязательно должна быть направлена на поддержку бронхолегочной системы, предупреждение осложнений от повторяющихся бронхитов и пневмоний. При развитии инфекционных заболеваний показано лечение антибиотиками. Обязательно используют методы и средства, которые способствуют разжижению мокроты и ее свободному удалению из бронхов и легких. Муколитики – препараты, разжижающие мокроту, используют длительными курсами, делая небольшие перерывы.
Диета при муковисцидозе – это не временные ограничения, а пожизненный образ питания. В рационе человека должно быть большое количество белка, получаемого из нежирного мяса, качественной рыбы, творога и яиц. Желательно, чтобы диета была высококалорийной, но необходимо ограничить количество поступающих жиров животного происхождения, трансжиров и грубой клетчатки, которая раздражает воспаленные стенки желудочно-кишечного тракта.
При развитии непереносимости лактозы из рациона исключают молоко. Из-за сухости слизистых оболочек и нарушения секреции слизи рекомендуется повышенный питьевой режим, особенно в жаркие месяцы.
Препарат Креон ® при муковисцидозе
Также в линейке Креон ® есть специальная форма – Креон ® Микро, выпущенная специально для лечения детей с муковисцидозом. Минимикросферы помещены во флакон россыпью, в комплекте идет мерная ложечка, которая позволяет удобно насыпать нужное количество препарата.
Подробнее о Креон ® Микро можно прочитать здесь.
Синдром Шегрена – что это такое? Причины, симптомы и лечение у опытных врачей медицинской Клиники МЕДСИ
Оглавление
Синдром Шегрена – аутоиммунное системное поражение соединительной ткани. Патология отличается тем, что в нее вовлечены железы внешней секреции (преимущественно слезные и слюнные). Вследствие развития заболевания появляется выраженная сухость кожи, носоглотки, глаз, рта, трахеи и влагалища. Также сокращается выработка пищеварительных ферментов. Патология может развиваться как самостоятельная или сопровождать склеродермию, дерматомиозит и другие заболевания. Лечение симптома Шегрена следует начинать после обнаружения первых же признаков.
Патоморфология
На раннем этапе в процесс вовлекаются мелкие протоки желез. При развитии заболевания железистая ткань атрофируется и замещается соединительной. Это приводит к нарушению функций пораженного органа. Нередко даже при отсутствии других выраженных симптомов синдрома Шегрена у пациентов отмечаются признаки воспаления слюнных желез.
Причины развития
Причины возникновения патологии в настоящий момент до конца не установлены.
Наиболее вероятной считают теорию о патологической реакции иммунной системы, которая развивается в ответ на повреждение клеток ретровирусом (герпесом, ВИЧ и др.). Как вирусы, так и клетки эпителия, измененные под их воздействием, воспринимаются иммунной системой человека как чужеродные. Иммунная система защищает организм и вырабатывает антитела. Это и приводит к разрушению тканей железы. Нередко синдром Шегрена передается по наследству, встречается у родителей и детей, у близнецов.
Спровоцировать развитие патологии могут следующие факторы (нередко их комбинации):
Симптомы синдрома Шегрена
Симптомы синдрома Шегрена во много зависят от причин заболевания, но всегда требуют устранения (лечения), так как существенно снижают уровень качества жизни пациента.
К основным железистым признакам относят:
К внежелезистым проявлениям патологии относят:
Нередко пациенты жалуются на повышенную чувствительность к ряду медикаментозных препаратов (нестероидным противовоспалительным средствам, антибиотикам и др.).
Если у вас появилась сыпь на теле, повысилась температура, пересыхают слизистые или обнаруживаются другие симптомы, и вы не знаете, что это, но хотите начать лечение, следует обратиться к специалисту: только он может провести диагностику и выявить синдром Шегрена или другую патологию.
Диагностика
Такие симптомы, как жжение и сухость глаз, например, не всегда свидетельствуют о синдроме Шегрена, но становятся причиной обращения к врачу с целью профилактики и лечения. Профессионалу очень важно точно распознать заболевание.
Диагностировать синдром Шегрена можно при наличии воспалительного процесса. Но в некоторых случаях воспаление провоцируется другими патологиями (например, сахарным диабетом). Для этого заболевания также характерно снижение секреции слюны. По этой причине диагностика должна быть максимально точной.
Наиболее информативным методом является биопсия слюнных и слезных желез с последующей гистологией полученного материала. Она проводится быстро и не доставляет пациентам выраженного дискомфорта. Фрагменты слизистых оболочек исследуются под микроскопом. Благодаря этому специалистам удается зафиксировать поражение желез.
Осложнения
К основным осложнениям синдрома Шегрена относят:
Патология прогрессирует как без лечения, так и в том случае, если терапия проводится неправильно. Именно поэтому следует обращаться только к высококвалифицированным врачам, располагающим опытом работы с пациентами с синдромом Шегрена.
Лечение заболевания
Основными задачами в терапии синдрома Шегрена являются снятие воспаления пораженных органов и устранение симптомов слизистых оболочек.
Для устранения воспалительного процесса назначаются:
В ходе подготовки к лечению пациентам проводят плазмаферез, позволяющий очистить кровь.
Для профилактики сухости конъюнктивы назначают препараты искусственной слезы и мази. Уход за полостью рта заключается в тщательном полоскании после каждого приема пищи.
Важно! Лечение синдрома Шегрена всегда проводится только под контролем врача-ревматолога.
Прогноз
Синдром Шегрена опасен тем, что может приводить к повреждению жизненно важных органов, постепенно прогрессировать. Бывают и случаи длительных ремиссий, когда патология никак не проявляет себя и больному кажется, что он полностью излечился, но болезнь внезапно возвращается. Одним пациентам помогает только симптоматическое лечение, другие долгое время борются с постоянным дискомфортом. Качество жизни многих больных существенно снижается. Пациенты страдают от суставных болей, сухости слизистых, упадка сил.
Важно! Пациенты с синдромом Шегрена подвержены высокому риску неходжкинской лимфомы. У некоторых больных развиваются другие онкологические заболевания, которые могут стать причиной не только снижения качества жизни, но и смерти.
При правильном и комплексном лечении пациенты могут рассчитывать на длительную и стойкую ремиссию. Но терапия должна быть комплексной и начаться как можно раньше – после обнаружения первых же признаков патологии.
Преимущества лечения синдрома Шегрена в МЕДСИ
Если вы хотите записаться на прием к ревматологу, позвоните
Что такое боковой амиотрофический склероз? Причины возникновения, диагностику и методы лечения разберем в статье доктора Калинкина М. Э., невролога со стажем в 8 лет.

Определение болезни. Причины заболевания
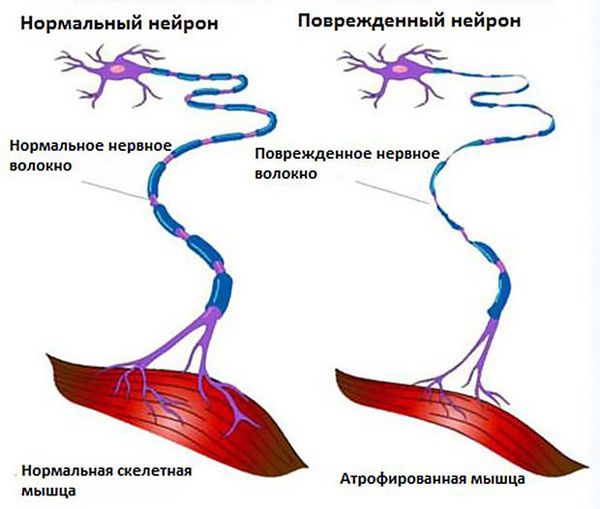
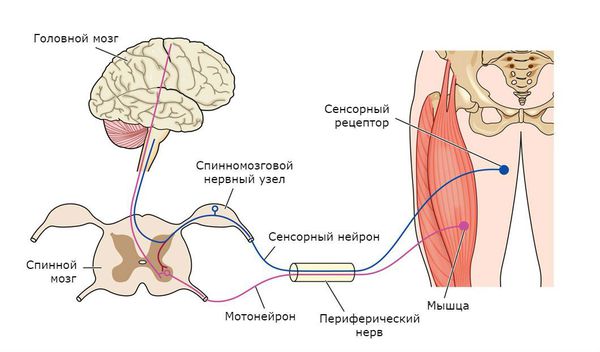
Чтобы понять суть заболевания, необходимо коснуться строения и функций головного и спинного мозга. В структуре спинного мозга на всём его протяжении и частично в стволе головного мозга существуют клетки, посылающие нервный импульс прямо к мышечным волокнам. Они называются нижние мотонейроны, так как своими импульсами приводят мышцы в движение. Группируясь в передней части поперечного среза спинного мозга, нижние мотонейроны образуют так называемый «передний рог».

Также спинной мозг выполняет функцию соединяющего нервного «кабеля» между головным мозгом и частями тела. В норме спинной мозг подчиняется головному мозгу. Это означает, что, если импульс от головного мозга укажет мышце поднять руку, а импульс спинного мозга укажет опустить, то рука поднимется.
Головой мозг имеет сложное строение, но в данной статье нужно уделить внимание верхним мотонейронам. Они группируются в области, которая отвечает на исходную генерацию двигательных импульсов в организме — коре больших полушарий и стволе мозга. Импульсы, в свою очередь, идут по спинному мозгу вниз к вышеупомянутым нижним мотонейронам, а оттуда к мышцам, приводя их в движение. Именно этот путь поражается при боковом амиотрофическом склерозе.

Боковой амиотрифический склероз — представитель группы болезней двигательного нейрона, т. е. болезней мотонейронов. Кроме БАС, в эту группу включены: первичный латеральный склероз, прогрессирующая мышечная атрофия и прогрессирующий бульбарный паралич.
В ходе многолетних исследований были найдены негенетические модифицируемые факторы образа жизни, предрасполагающие к БАС:
Обобщая вышесказанное, можно сказать, что БАС развивается в результате комбинированного воздействия генов, факторов окружающей среды и образа жизни. Эта модель (ген-время-среда) предполагает, что развитие БАС является многоэтапным процессом, в котором генетические дефекты являются лишь одним из нескольких этапов, в конечном итоге приводящих к БАС.
Симптомы бокового амиотрофического склероза
Начальные симптомы БАС варьируют у пациентов в зависимости от степени поражения верхних и нижних двигательных нейронов и от того, какие участки тела вовлечены. Поэтому особенность бокового амиотрофического склероза заключается в том, что врачи часто вынуждены наблюдать за развитием жалоб в течение нескольких месяцев, прежде чем поставить верный диагноз (например, определить тип заболевания двигательного нейрона или тип БАС)
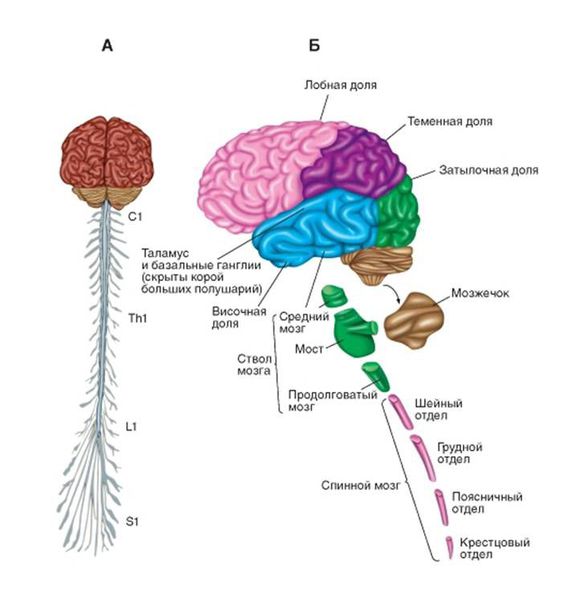
| Уровень поражения нижнего мотонейрона | Ствол головного мозга | Шейный отдел спинного мозга | Грудной отдел спинного мозга | Поясничный отдел спинного мозга |
|---|---|---|---|---|
| Группы мышц, которые поражаются | Мыщцы лица, мягкого нёба, язык, мышцы гортани и глотки | Мышцы шеи, рук | Мышцы спины, живота, диафрагмы | Мышцы спины, живота, ног |
По мере развития заболевания будут наблюдаться признаки поражения верхнего мотонейрона, а также постепенное присоединение соседних отделов спинного мозга.
Признаки поражения верхнего мотонейрона:

Патогенез бокового амиотрофического склероза
Патогенез заболевания чётко не определён. Примерная схема такова: ген в хромосоме (например ген супероксиддисмутазы-1, находящийся на 21-й хромосоме) «ломается» по неизвестным причинам. В результате синтезируются неправильные белки, которые мешают нормальной работе клеток нервной системы. Нервные клетки из-за этой поломки погибают, но процесс переработки мёртвых клеток (процесс аутофагии) также по какой-то причине не происходит. Из-за этого мёртвые клетки не распадутся на составляющие, а значит не создают «строительного материала» для новых клеток.
Все это соотносится с клиникой. Повышенная активность здоровых нейронов вызывает подёргивание мышц. Вскоре из-за скопления мёртвых клеток и отравления глутаматом нейроны погибают, происходит денервация мышц (разобщение связей мышц с нервной системой), в результате чего те слабеют и истончаются.
Классификация и стадии развития бокового амиотрофического склероза
Единой классификации БАС не существует, поскольку не существует единства представлений о его патогенезе. Рассмотрим Североамериканскую классификацию, которая близка к отечественной. Согласно этой классификация, БАС делится на две большие группы в зависимости от причин: семейный и спорадический (случайный).
Спорадический БАС делится на группы относительно уровня поражения моторных нейронов двигательного пути в дебюте заболевания. БАС может впервые проявиться на уровне ствола мозга, шейного, грудного (в том числе в виде слабости диафрагмальных мышц), поясничного отдела спинного мозга или на всем протяжении пути единовременно.
Семейная группа делится на виды в зависимости от типа мутации конкретного гена супероксиддисмутазы-1 (СОД-1) или мутаций в других хромосомах (около 10 других известных генов).
Североамериканская классификация БАС
Спорадический (случайный) БАС
В первую группу кроме классического БАС входят три патологии: первичный латеральный склероз; прогрессирующая мышечная атрофия; прогрессирующий бульбарный паралич. В нашей стране их считают отдельными заболеваниями и объединяют в группу болезней двигательного нейрона. В США данные патологии рассматривают как варианты проявления БАС. Несмотря на разницу классификаций, проявления этих заболеваний будут одинаковыми в любой стране.
Осложнения бокового амиотрофического склероза
Диагностика бокового амиотрофического склероза
При подозрении на болезнь двигательного нейрона пациент сначала отвечает на стандартные вопросы невролога о жалобах, развитии болезни, сопутствующих заболеваниях (стоит обратить внимание на онкологию и инфекции, так как они могут стать причиной ложноположительного БАС-симптома, а не самой болезни). Врач также спрашивает о болезнях родственников, стране рождения, о мочеиспускании и дефекации.
Невролог должен осмотреть мышцы тела, оценить их силу, тонус, проверить рефлексы с помощью молоточка, оценить чувствительность кожи при помощи иголки и камертона. При подозрении на БАС мышцы непроизвольно сокращаются и истончаются. В них теряется сила, даже если пациент занимается физическими упражнениями для их укрепления. Также врач может задать вопросы, похожие на определение IQ. При снижении интеллектуальных способностей можно заподозрить некий патологический процесс в коре головного мозга, где находится верхний мотонейрон.
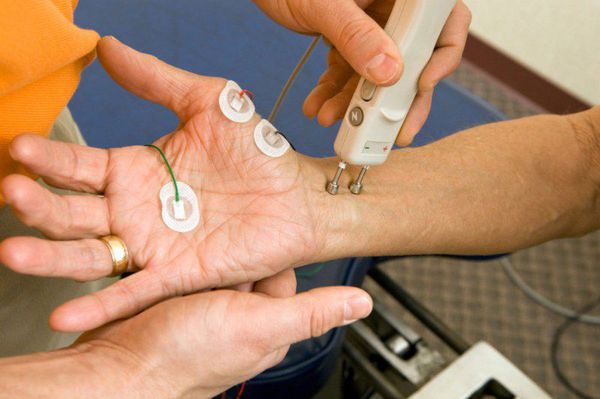
Диагноз бокового амиотрофического склероза невозможно достоверно поставить с первого посещения невролога только после одного исследования ЭМГ. Иногда с целью уточнения заболевания или формы БАС врач дополнительно может назначить:
Функциональную способность пациентов оценивают в баллах по специальной шкале. В соответствии с этой шкалой максимальный балл 48 соответствует полной функциональной способности больного, а минимальный балл 0 соответствует максимально выраженной инвалидизации пациента.
Лечение бокового амиотрофического склероза
Препарат эдаравон является поглотителем свободных радикалов. Это внутривенное лекарственное средство для лечения БАС, которое было одобрено агентством Министерства здравоохранения США в мае 2017 года (примерно через 2 года после его одобрения для лечения БАС в Японии и Южной Корее). Использование этого препарата было оценено при разных неврологических расстройствах, включая БАС, в нескольких клинических испытаниях. Результаты были примечательными. Отмечалось замедление снижения показателей функциональной шкалы на 33 % за 6 месяцев и замедление ухудшения качества жизни людей с БАС в целом. Относительно расстройств дыхания не удалось достичь статистически значимых результатов.
Прогноз. Профилактика
Профилактика заключается в исключении или снижении влияния факторов риска: необходимо отказаться от курения; не контактировать с вредными веществами (пестицидами, гербицидами, свинцом и др.); исключить микротравмы головы.
Появляются и другие исследования, опровергающие пользу «витаминотерапии» для стран, не испытывающих проблем голода и нехватки пищи среди населения. В связи с этим более целесообразно рекомендовать пациентам покупать продукты «средиземноморской диеты», т. е. употреблять в пищу естественные витамины, антиоксиданты, жирные кислоты, не опасаясь передозировки этих веществ. Жителям отдалённых регионов с материальной точки зрения иногда выгоднее покупать пищевые добавки для непродолжительного курса приёма, чем продукты средиземноморской диеты.
Спинальная мышечная атрофия 1 типа
Может ли донор банка половых клеток оказаться носителем тяжелого генетического заболевания?
У людей, решивших воспользоваться услугами банка половых клеток, обычно возникает множество вопросов и переживаний. Часто пара беспокоится о том, что донор может оказаться носителем серьезных наследственных болезней. Например, многие слышали, что есть такая тяжелая болезнь, как спинальная мышечная атрофия. Поговорим о том, что это за патология, и как в Репробанке выявляют кандидатов в доноры, являющихся ее носителями.
Спинальная мышечная атрофиия 1 типа, или младенческая форма СМА (англ. SMA), или болезнь Верднига-Гоффмана – тяжелое наследственное заболевание, которое проявляется в нарушении работы мышц, дыхания, глотания.
Заболевание развивается из-за дефекта в гене SMN1, который находится в длинном плече пятой хромосомы. В нём возникает делеция – выпадение участка генетического материала, которая приводит к некорректной работе гена и отсутствию выработки белка, отвечающего за поддержание двигательных нейронов, являющихся нервными клетками головного и спинного мозга, отвечающими за управление движением мышц.
Спинальная мышечная атрофия 1 типа наследуется аутосомно-рецессивно. То есть для того, чтобы родился больной ребенок, он должен получить генетический дефект сразу в двух копиях гена SMN 1, расположенных на сестринских хроматидах, пришедших от обоих родителей.
Если это происходит, то в организме не синтезируется белок SMN, необходимый для нормальной работы двигательных нейронов (нервных клеток), из-за чего возникают мышечные расстройства.
Внимательный читатель тут задаст вопрос: коль скоро существует первый тип спинальной мышечной атрофии, то должен быть второй и т. д.? Действительно, всего существует пять типов заболевания. Они различаются по характеру генетических нарушений, времени возникновения и тяжести симптомов. Первый тип самый распространенный и один из самых тяжелых.
Частота носительства дефектных генов оценивается, по разным данным, от 1:38 до 1:70. Иными словами, каждый 38–70-й человек имеет одну дефектную копию гена SMN1, на одной из дочерних хроматид, но сам при этом здоров. Если два таких человека решат завести ребенка, вероятность того, что малыш получит оба дефектных гена и родится больным, составляет 25%. Но, как считается, такая тяжелая патология нередко приводит к прерыванию беременности на ранних сроках, поэтому распространенность заболевания не так высока, как должна бы получиться по расчетам: в реальности она составляет 1 случай на 11 000 новорожденных.

Как проявляется младенческая форма СМА?
Симптомы спинальной мышечной атрофии возникают с рождения либо до 6 месяцев. У таких детей отмечается гипотония – сильное ослабление мышечного тонуса, особенно в ручках и ножках. Из-за поражения нервных клеток их мышцы ослаблены, поэтому у таких малышей не формируются основные навыки: они не могут держать головку, сидеть, ползать, стоять. У них нарушено дыхание и глотание, утеряна способность самостоятельно принимать пищу, и их приходится кормить через зонд. Такие дети нуждаются в специальном уходе с участием команды врачей-специалистов.
При этом когнитивные (познавательные) способности больного ребенка обычно не нарушены.
Для выявления болезни Верднига-Гоффмана проводят электромиографию (регистрацию электрических импульсов в мышцах), биопсию мышц (получают небольшой образец мышечной ткани и исследуют под микроскопом). Самый точный метод диагностики – исследование структуры ДНК (для анализа берут кровь).
Каков прогноз? Существует ли эффективное лечение?
Прогноз при спинальной мышечной атрофии неблагоприятный. Большинство детей, родившихся с этим заболеванием, не доживает до 2 лет. Средняя продолжительность жизни составляет 1 год.
Лечения, направленного на причину заболевания, на данный момент не существует, хотя предпринимаются попытки его создать. Большинство детей получают лишь соответствующий уход и симптоматическую терапию. Симптомы постепенно нарастают, и ребенок погибает, как правило, от дыхательной недостаточности.
В 2016 году американским Управлением по контролю качества лекарственных препаратов и продуктов питания (FDA) был одобрен препарат Спинраза (Spinraza), модифицирующий течение спинальной мышечной атрофии. Дело в том, что помимо гена SMN1, в клетках человека есть ген SMN2, который тоже кодирует белок SNM, но его вырабатывается меньше, и он быстрее разрушается. Спинраза заставляет ген SMN2 работать активнее, в результате чего улучшается состояние больного ребенка и увеличивается продолжительность жизни.
В 2019 году FDA одобрило инновационный препарат для генной терапии спинальной мышечной атрофии – Золгенсма (Zolgensma). Это лекарство – по сути искусственно созданный «вирус», который доставляет рабочую копию гена SMN1 в нервные клетки.
В августе 2019 года состоялась регистрация препарата Спинраза (Spinraza ) в РФ.
Несмотря на эти достижения, до победы над СМА еще далеко, а инновационные препараты стоят очень дорого, и к сожалению, доступны не многим, болеющим СМА детям. Поэтому врачи Репробанка тщательно обследуют кандидатов в доноры половых клеток и информируют пациентов банка о необходимости уточнения их статуса носительства частых аутосомно-рецессивных заболеваний, к которым относится СМА, что позволяет значительно снизить риски рождения детей с генетической патологией.
Какие меры принимаются в Репробанке?
Репробанк тщательно проверяет всех доноров половых клеток:
Пользуясь услугами Репробанка, вы максимально снижаете риски передачи наследственных заболеваний от доноров репродуктивных клеток.
Существуют некоторые причины для остаточного риска рождения ребенка с заболеванием, но они крайне малы:
Все эти ситуации встречаются достаточно редко. Для того чтобы дополнительно снизить риски, мы рекомендуем провести генетический анализ на данную патологию второму родителю.

Зиновьева Юлия Михайловна
Ведёт генетическое обследование доноров Репробанка, осуществляет подбор доноров для пар, имеющих ранее рождённых детей с установленной генетической патологией.



