Auc в медицине это что
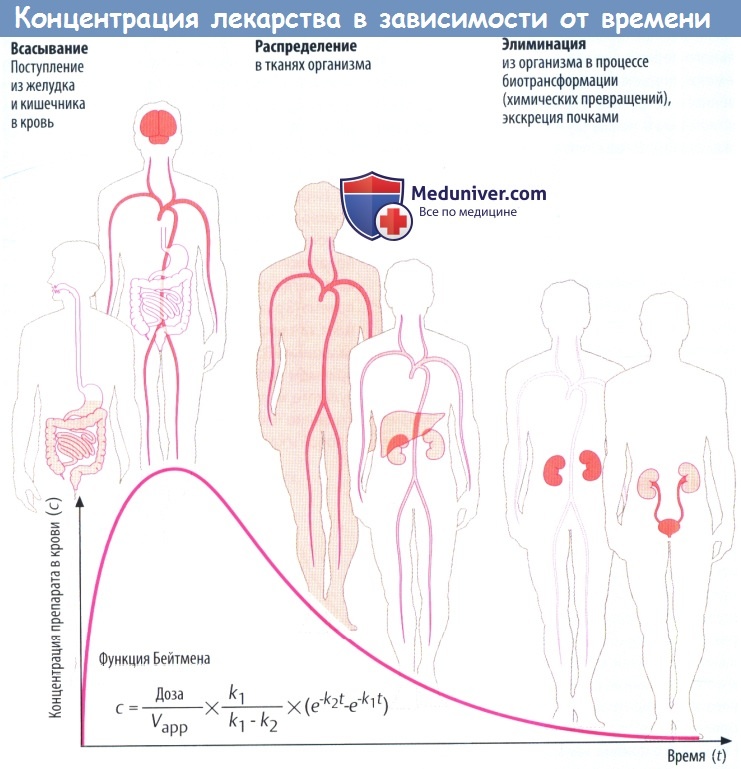
(А) Лекарственные средства попадают в организм и выводятся из него разными путями. Таким образом, организм представляет собой открытую систему, в которой фактическая концентрация препарата отражает взаимодействие между его поступлением (приемом) и эвакуацией (элиминацией).
Скорость всасывания препарата в желудке и кишечнике зависит от множества факторов: скорости растворения вещества (в случае приема твердой лекарственной формы) и транзита по ЖКТ, проницаемости слизистой для препарата, его градиента концентрации на границе слизистой и крови,кровоснабжения слизистой оболочки.
Всасывание из кишечника приводит к повышению концентрации лекарственного вещества в крови. Препарат разносится с кровью к различным органам (распределение), которые поглощают его в количестве, соответствующем его химическим свойствам и скорости кровотока через орган.
Например, органы с хорошим кровоснабжением, такие как головной мозг, получают большее количество препарата, чем органы с низким кровоснабжением. В результате поглощения тканями происходит снижение концентрации лекарственного вещества в крови. По мере снижения градиента на границе слизистой оболочки и крови всасывание в кишечнике замедляется. Пик концентрации в крови достигается тогда, когда количество вещества, покидающего кровь за единицу времени, равно количеству всосавшегося.
Поступление вещества в ткани печени и почек представляет собой перемещение в органы выведения. Концентрация препарата в крови в различные периоды времени представляет собой совокупность процессов абсорбции, распределения и элиминации, которые пересекаются во времени.
Если распределение происходит значительно быстрее, чем элиминация, снижение концентрации в крови вначале происходит быстро, а затем замедляется. Фаза быстрого снижения обозначается как α-фаза (фаза распределения), медленного — как β-фаза (фаза элиминации). Если препарат распределяется быстрее, чем абсорбируется, концентрацию препарата в крови можно описать математически упрощенной функцией Бейтмена (k1 и k2 — константы скорости для абсорбции и элиминации соответственно).
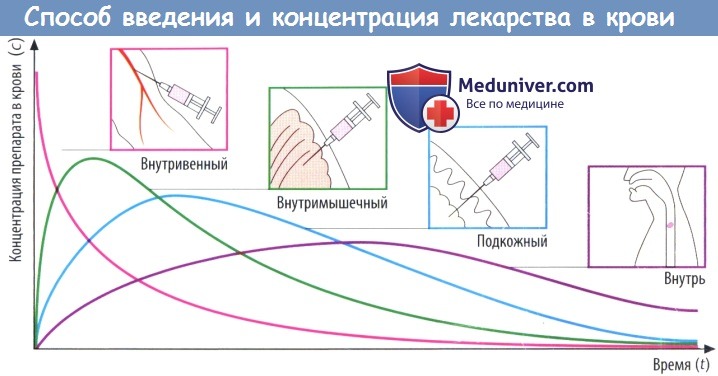
(В) Скорость абсорбции зависит от способа введения препарата. Чем выше скорость абсорбции, тем короче будет время (tmax), которое требуется для достижения пика концентрации в плазме (cmax), тем выше будет cmax и тем раньше уровень препарата в крови снова начнет снижаться.
Площадь под кривой, описывающей зависимость концентрации препарата в крови от времени (AUC), не зависит от пути введения препарата при условии, что доза и биодоступность остаются теми же (закон соответственных состояний). Таким образом, AUC можно использовать для вычисления биодоступности (F) препарата.
Значение AUC, измеренное после приема внутрь и в/в введения определенной дозы конкретного лекарственного вещества, соответствует проценту вещества, попавшего в системный кровоток после приема внутрь: F = AUCприем внутрь/AUCв/в введение.
Определение концентрации препарата в крови позволяет сравнить различные патентованные лекарственные средства, содержащие одно и то же действующее вещество в одинаковой дозе. Идентичные кривые зависимости концентрации в крови от времени для препаратов различных производителей (при условии стандартных лекарственных форм) означают биоэквивалентность стандартного вещества и нового исследуемого препарата.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Auc в медицине это что
Доза химиопрепаратов обычно рассчитывается в мг/кг массы тела или мг/м 2 всей площади поверхности тела. Расчет дозы, основанный на площади поверхности тела, предпочтительнее основанного на массе тела, т. к. площадь поверхности изменяется гораздо меньше, позволяя более точно рассчитать дозу препарата на протяжении всего курса лечения.
Расчет дозы посредством этой единицы измерения также более сопоставим для взрослых и детей, и различия в общей дозе между очень полными и худыми людьми минимальны. Расчет дозы для экспериментальных животных выраженный в мг/м2 более доступен для перерасчета у человека.
Для взрослых дозы в мг/кг можно преобразовать с приемлемой точностью в мг/м 2 умножением на 40.

Необходимо корректировать дозы препаратов химиотерапии (XT) для больных, у которых, вероятно, имеются нарушения функции костного мозга; это пациентки старше 70 лет и те, кому ранее проводилась тазовая или абдоминальная лучевая терапия (ЛТ) либо химиотерапия (XT).
Корректировка дозы часто требуется для больных, получающих противоопухолевые препараты, которые выводятся почками. Это уменьшает вероятность слишком высокой концентрации препаратов в плазме и сопутствующего риска серьезных нарушений функции почек. Некоторые методы, используемые для оценки функции почек (скорость клубочковой фильтрации — СКФ) индивидуальны для каждой конкретной опухоли.
Наиболее часто применяется расчет клиренса креатинина (КК), использующего показатели сывороточного креатина. Элиминация креатинина происходит главным образом с помощью клубочковой фильтрации, хотя небольшое количество может выделяться через почечные канальцы. Некоторые исследования сравнили различные методы оценки КК, используя оценку сывороточного креатинина. Эти методы основываются на корреляции КК с возрастом, массой тела, сывороточным креатинином и его метаболизмом. Наиболее используемые методы описаны ниже.
Метод Джеллиффа для расчета клиренса креатинина (КК)
Изначально метод Джеллиффа применялся в качестве простой оценки КК на основании уровня сывороточного креатинина, делая незначительные изменения в расчетах в соответствии с полом больного.
Современная формула Джеллиффа принимает во внимание возраст и функцию почек:
КК(мл/мин)= 1,73 [(100/сывороточный креатинин в мг/дл) — 2].
Ее используют в 90 % случаев для оценки КК у женщин.
Метод Кокрофта—Голта для расчета клиренса креатинина (КК)
Это уравнение включает показатели для сухой массы тела (без жира), которые особенно важны для полных больных (полученное значение умножается на 0,85 для женщин).
Этот метод сходен с расчетом по Джеллиффу и представляет собой следующее:
КК = (140 — возраст) х (сухая масса тела в кг)/(сывороточный креатинин в мг/дл) х 72.
Формула Калверта для расчета дозы химиопрепарата
Использование КК также было включено в так называемую формулу Калверта. Метод основан на надежных показателях; имеются данные, показывающее, что существует обратная линейная корреляция между СКФ и AUG у таких препаратов, как карбоплатин.
Для того чтобы получить желаемую AUC, необходимо не только снизить дозу препарата для больных с почечной недостаточностью, но и увеличить стандартные дозы для больных с высокими показателями почечного клиренса.
Формула Калверта:
Доза (мг) = AUC х (СКФ + 25).
Первоначально вычисления по Калверту проводились по СКФ, измеренной с помощью хром-51-ЭДТА метода. Хотя КК может превысить значение СКФ на 10—40 %, большинство врачей вычисляют СКФ, используя формулы Джеллиффа или Кокрофта—Голта, и затем подставляют эти значения в формулу Калверта.
Схема расчета индивидуальной дозы карбопластина:
1. Доза карбоплатина (мг) = AUC х (СКФ + 25)
2. AUC выбрана для соответствующих клинических ситуаций:
— AUC 6 для нелеченных больных, когда использованы комбинации с таксанами
— AUC 5 для ранее леченных больных
— AUC 7 для первично не леченных больных
3. СКФ служит эквивалентом КК, которую можно измерить или оценить в зависимости от роста, массы тела и уровня сывороточного креатинина


Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Критерии точности дерматоскопической диагностики: от виртуальных процентов к реальным цифрам
В чем измеряется точность диагностики при дерматоскопическом исследовании кожи с целью выявления злокачественных новообразований? Ответить на этот вопрос не так легко, как может показаться на первый взгляд. Возможно, вы сталкивались с примерно такими заявлениями: «метод А на столько-то процентов точнее метода В». И все бы ничего, но какая точность первого метода? Об этом многие авторы предпочитают тактично умалчивать.
Между тем, от точности диагностики напрямую зависит здоровье и жизнь пациента – например, если речь идет о выявлении злокачественных новообразований кожи. Это большая проблема сегодня не только в России, но и во всем мире.
По данным Московского научно-исследовательского онкологического института имени П.А. Герцена, в 2018 году в Российской Федерации впервые в жизни выявлено 624 709 случаев злокачественных новообразований (ЗНО) – в том числе 285 949 у мужчин и 338 760 у женщин. На учет были взяты 542 569 «новых» пациентов.
В целом же показатель заболеваемости ЗНО в России составляет 425,4 на 100 000 населения – это на 1,2% выше 2017 года и на 23,1% выше 2008 года. Удельный вес конкретных злокачественных патологий представлен в таблице 1.
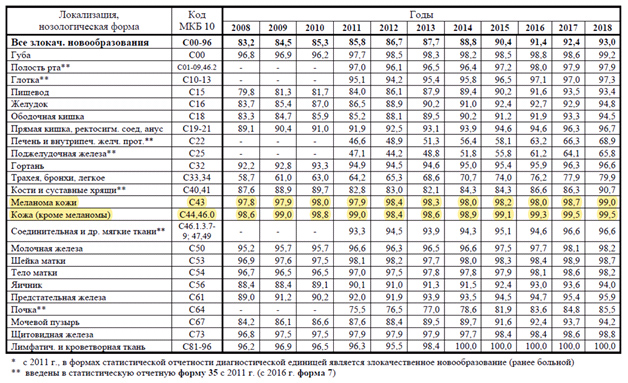
Табл. 1. Удельный вес больных с морфологически подтвержденным диагнозом относительно всех пациентов с впервые в жизни установленным диагнозом ЗНО в России в 2008-2018 годах, %* (Состояние онкологической помощи населению России в 2018 году. Под ред. А.Д. Каприна, В.В. Старинского, Г.В. Петровой — М.: МНИОИ им. П.А. Герцена — филиал ФГБУ «НМИЦ радиологии» Минздрава России, 2019. — илл. — 236 с)
Дерматоскопия является основным методом раннего выявления меланомы. В статье мы разберемся, как именно оценивается ее точность, и какие варианты диагностики существуют сегодня.
Нерешенные проблемы
Дерматоскопия представляет собой неинвазивный метод визуальной оценки поражений кожи и скальпа. С его помощью врач может распознать морфологические структуры, незаметные при «невооруженном» осмотре, и правильнее оценить клинические и морфологические изменения кожи, особенно пигментных поражений.
Распространенной ошибкой, которую многие врачи допускают случайно или преднамеренно, является ограничение дермоскопической оценки только подозрительными поражениями. С одной стороны, это помогает сэкономить время на рутинном приеме, но с другой, все имеющиеся меланоцитарные невусы должны быть изучены, вне зависимости от их локации и размера.
Современные исследования говорят о том, что меланомы чаще диагностируются среди небольших меланоцитарных поражений (размером менее 3 мм) по сравнению с более крупными, которые вызывают гораздо больше беспокойства у пациентов (Bono A., et al. Micro-melanoma detection: a clinical study on 206 consecutive cases of pigmented skin lesions with a diameter 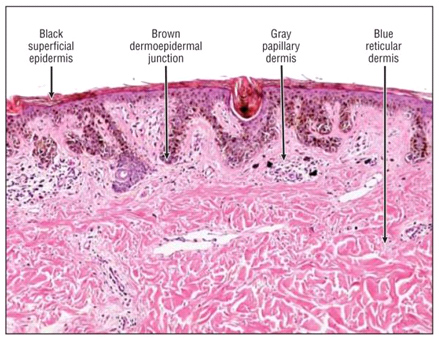
Рис. 2. Влияние цвета новообразования при дерматоскопии на глубину его залегания: черный – поверхностно в эпидермисе, коричневый – в дермоэпидермальном соединении, серый – в папиллярной дерме, голубой – в ретикулярной дерме (Zalaudek I., et al. Using dermoscopic criteria and patient-related factors for the management of pigmented melanocytic nevi. Arch Dermatol 2009; 145: 816-826)
Интересно, что клиническое обследование пигментных поражений диаметром менее 3 мм с подозрением на меланому показывает среднюю чувствительность 43% и специфичность 91%. В свою очередь, дерматоскопия показывает гораздо более высокую чувствительность (83%) и низкую специфичность (69%).
В этот момент мы вплотную подошли к понятию точности диагностики. Но сперва нужно отметить, что все вышеназванные правила (ABCDE и другие) являются «правилами» лишь для людей. Врачи выработали их за многие годы работы, но если вместо человека поставить машину – достаточно мощный компьютер с искусственным интеллектом, он будет оценивать опухоли по собственным критериям. Об этом чуть ниже.
Чувствительность и специфичность
Одной из важнейших задач дерматолога является раннее выявление меланом. От точности его диагностики во многом зависит здоровье и жизнь пациента. Дерматоскоп не может самостоятельно поставить диагноз и даже предположить наличие злокачественного новообразования, поэтому врач является его неотъемлемой составляющей. Опыт, навыки и внимательность специалиста во многом определяют точность диагностики, которая выражается в ее чувствительности и специфичности.
Чувствительность диагностики – это отношение выявленных заболевших людей к общему числу обследованных заболевших.
Допустим, у нас есть 5 пациентов: 1 и 4 здоровы, а у 2, 3 и 5 – меланома (рис. 3). Несколько врачей провели дерматоскопию и получили следующие результаты:

Рис. 3. Пятеро пациентов: улыбающиеся здоровы, грустные заболели
Как видите, чувствительность дерматоскопии, как и любого другого метода диагностики, зависит от правильности «угадывания» реально заболевших людей. Здоровым людям также можно ставить диагноз, хотя на самом деле это ошибка, ведь у них его нет. Но на показатель чувствительности это не влияет – ее суть заключается в выявлении заболевших людей, как говорится, любой ценой. Поэтому нельзя рассчитывать только на эту характеристику метода. Ведь чувствительность первого и четвертого врача одинакова (100%), но качество их диагностики абсолютно разное. И за это качество отвечает другой параметр – специфичность.
Специфичность диагностики – это отношение выявленных здоровых людей к общему числу обследованных здоровых.
Вернемся к пациентам с рисунка 3 и оценим специфичность каждого из врачей:
Идея специфичности заключается снижении числа ложноположительных диагнозов, когда за меланому принимают обычный невус. Любое удаление новообразования – не важно, доброкачественное оно или злокачественное, является травмирующей операцией с определенными рисками. К тому же это влечет дополнительные затраты для пациента и государства на хирургию.
Четвертый врач оказался самым точным – он верно обнаружил всех здоровых людей, поэтому его специфичность составила идеальные 100%. Этот специалист большой молодец, ведь его чувствительность также равна 100%, и нам всем нужно стремиться к такой точности!
К сожалению, в реальной жизни дела обстоят несколько иначе:
Это происходит потому, что чувствительность, образно говоря, отвечает за скорость постановки диагноза, а специфичность – за внимательность. И чем более внимательно врач будет рассматривать каждый невус, тем больше времени это займет. Напротив, если он быстро поставит всем диагноз меланомы, это почти наверняка не будет соответствовать действительности. Хотя поступая так, врач, безусловно, «угадает» и реально больных пациентов.
В поисках идеального алгоритма
Какова реальная точность врачебной диагностики? Для ответа на этот вопрос немецкими специалистами было проведено любопытное исследование.
Возможно, вы слышали о сравнении навыков Человека и Машины при игре в шахматы. В далеком 1997 году разработанный компанией IBM искусственный интеллект Deep Blue обыграл легендарного чемпиона Гарри Каспарова со счетом 4:2. Хотя эта победа была достигнута перебором вариантов, она символизировала безграничные возможности зарождавшегося компьютерного разума. С тех пор прошло более 20 лет, искусственный интеллект стал намного сложнее и научился помогать людям в поиске злокачественных новообразований.
Немецкие ученые сравнили точность диагностики современного искусственного интеллекта (ИИ) и врачей-дерматологов со всего мира. Для этого был создан Тестовый набор-300, который включал в себя 300 дерматоскопий: 20% из них являлись подтвержденными меланомами с различных областей тела, а остальные 80% – доброкачественными меланоцитарными невусами.
К участию были приглашены дерматологи со всего мира, откликнулись 58 человек из 17 стран. Из них 17 специалистов были начинающими (менее 2 лет стажа), 11 – опытными (2-5 лет) и 30 экспертами (более 5 лет в профессии). Сперва им выслали 100 простых снимков невусов из Тестового набора-300, глядя на которые врачам следовало определиться с тактикой: удалить, наблюдать или ничего не предпринимать (Level 1). Через месяц им прислали 100 уточненных снимков, с дерматоскопией и клинической картиной – врачам предстояло вновь определиться с тактикой (Level 2).
Оппонентом людям выступал искусственный интеллект – сверточная нейронная сеть на основе архитектуры Google V4. Это особый тип ИИ, созданный исключительно для анализа изображений. Его особенность заключается в раскладывании каждого снимка на множество слоев с их детальным анализом (рис. 4). Предварительно он был натренирован на 100 тысячах дерматоскопий с клинически подтвержденным диагнозом, а сейчас ему предстояло показать себя в деле. Для ИИ были взяты оставшиеся 100 снимков, а в качестве дополнительной работы машина проанализировала весь Тестовый набор-300.
Рис. 4. Раскладка изображения на слои в процессе анализа сверточной нейронной сетью (Made S.W., et al. Abnormal heart rhythm detection based on spectrogram of heart sound using Convolutional Neural Network. CITSM 2018; 1-4)
Результаты врачей и машины сравнивались на графике Receiver Operating Characteristics (ROC) – Рабочая характеристика приемника. Он показывает соотношение между долей объектов с определенным признаком, верно классифицированных как имевшие этот признак, и долей объектов без признака, ошибочно названных имевшими признак. Проще говоря, ROC-кривая показывает, насколько часто врачи и машина верно угадывали больных с меланомой и насколько редко они ставили этот диагноз здоровым людям.
Поскольку точность диагностики у дерматологов разная, нужно было как-то объединить результат и вывести на графике среднее значение – за это отвечает Area Under The Curve (AUC) или Площадь под кривой. Она показывает усредненную точность врачебной диагностики – результат представлен на рисунке 5.
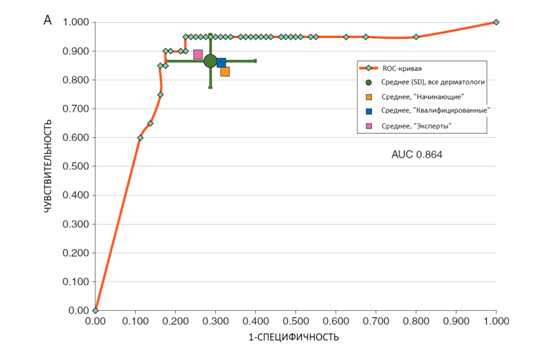
Рис. 5. Средняя точность врачебной диагностики в виде Площади под кривой (AUC). Как видно, эксперты оказались наиболее точными в поиске меланомы по сравнению с опытными и начинающими специалистами (Haenssle H. A., et al. Man against machine: diagnostic performance of a deep learning convolutional neural network for dermoscopic melanoma recognition in comparison to 58 dermatologists. Ann Oncol 2018; 29(8): 1836-1842)
Эксперты со стажем более 5 лет оказались наиболее точными – чувствительность 89%, специфичность 74,5%. У опытных врачей эти показатели были 85,9% и 68,5%, а у начинающих 82,9% и 67,6% соответственно (Level 1). Площадь под кривой (ROC AUC) для людей в Level 1 составила 0,79 (усредненное значение).
Повторный осмотр изображений с клинической картиной и дерматоскопиями (Level 2) почти не повлиял на чувствительность экспертов (89,5%), зато сильно поднял ее у опытных (90,9%) и начинающих врачей (86,6%). Специфичность также выросла у всех групп – примерно до 77-78%. Площадь под кривой (ROC AUC) для людей в Level 2 составила 0,82 (усредненное значение).
Что касается искусственного интеллекта, то на выборке из 100 снимков (Level 1) его чувствительность составила 95%, специфичность – 63,8%, ROC AUC Level 1 – 0,86. При анализе всего Тестового набора-300 (Level 2) чувствительность ИИ составила 95%, специфичность – 80%, ROC AUC Level 2 – 0,953 (рис. 6).
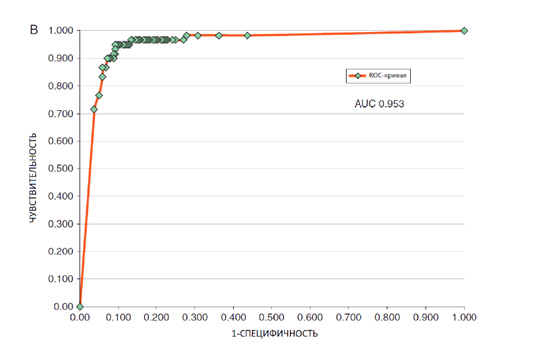
Рис. 6. Точность диагностики сверточной нейронной сети на базе архитектуры Google V4 при анализе Тестового набора-300 (Haenssle H.A., et al. Man against machine: diagnostic performance of a deep learning convolutional neural network for dermoscopic melanoma recognition in comparison to 58 dermatologists. Ann Oncol 2018; 29(8): 1836-1842)
В итоге искусственный интеллект в данном исследовании обошел врачей в точности диагностики на обоих уровнях. Причем алгоритм ИИ не занимается простым сравнением плохого с хорошим – он послойно разбирает каждый снимок и детально анализирует его, используя собственные критерии злокачественности новообразований и постоянно совершенствуя их.
Значит ли это, что можно избавить специалистов от рутинных обследований и оставить в диагностике меланомы компьютеры с операторами? В один прекрасный день это, безусловно, случится. Однако сейчас задача искусственного интеллекта заключается в то, чтобы помогать живому интеллекту (то есть врачу), повышая его чувствительность и специфичность.
Эра FotoFinder
Как вы поняли, основная проблема любого врача заключается в падении специфичности при росте чувствительности, и наоборот. Это происходит по разным причинам – не последнюю роль играет снижение концентрации, усталость и «замыливание» глаз при длительной работе. Искусственный интеллект призван решить эту проблему: ему не нужна еда, он не отвлекается на посторонние дела и никогда не устает – точность его диагностики одинаково высока 24 часа в сутки, 7 дней в неделю. Более того, из года в год она неизменно растет.
Иметь такого помощника станет большим подспорьем для любого специалиста, причем обзавестись собственным ИИ можно уже сегодня. Сверточная нейронная сеть на базе архитектуры Google V4 реализована в диагностическом комплексе FotoFinder (FotoFinder Systems GmbH, Германия).
Состав типичного комплекса FotoFinder:
FotoFinder не занимает много места – камера, штатив и указатель очень компактные, а компьютер с видеодерматоскопом легко помещаются на любом столе. Но в этом «хрупком теле» скрыт недюжинный интеллект. Ученые обучили его на 100 000 дерматоскопий с клинически подтвержденным диагнозом, и после старта работы он начал выявлять собственные критерии злокачественности опухолей. Да, FotoFinder не занимается простым сравнением плохого с хорошим, он внимательно анализирует каждый невус и буквально раскладывает его на атомы.
С помощью FotoFinder в 2016 году была найдена самая маленькая меланома в мире – ее диаметр всего 0,9 мм при дерматоскопии и 0,6 мм при гистологии. Искусственный интеллект помог человеку выявить мельчайшую злокачественную опухоль и спас жизнь пациенту. При этом внешне такая опухоль не привлекала внимания, и пропустить ее не составляло труда (рис. 7). Ее обнаружили при помощи картирования всего тела на комплексе FotoFinder ATBM bodystudio. В нем пациенту делают 20 стандартных снимков всего тела в разных позах, после чего программа детально анализирует каждый миллиметр его кожи.
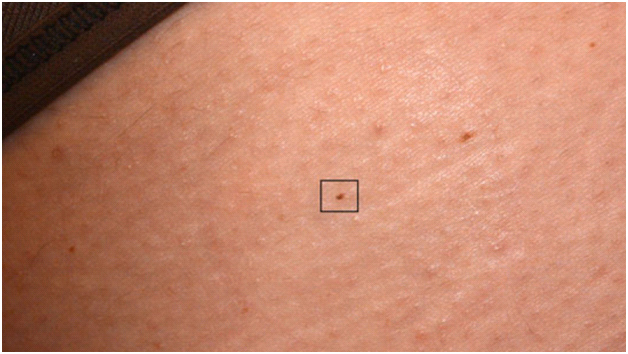
Рис. 7. Самая маленькая меланома в мире (d = 0,9/0,6 мм), выявленная искусственным интеллектом FotoFinder в 2016 г.
Таким образом, точность диагностики – это сочетание высокой чувствительности и высокой специфичности. Наглядным отражением точности является площадь под кривой (AUC) на графике рабочей характеристики приемника (ROC). Современные исследования показывают, что искусственный интеллект дает бо́льшую ROC AUC по сравнению с врачами-дерматологами – 0,86 против 0,79. Это делает его уникальным и незаменимым помощником в ежедневной практике для специалиста любого уровня.
Auc в медицине это что
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ И СОЦИАЛЬНОГО РАЗВИТИЯ
и социального развития
10 августа 2004 года
ПРОВЕДЕНИЕ КАЧЕСТВЕННЫХ ИССЛЕДОВАНИЙ
БИОЭКВИВАЛЕНТНОСТИ ЛЕКАРСТВЕННЫХ СРЕДСТВ
— время» в интервале времени от 0 до момента (t) отбора последней
— время» в интервале времени от 0 до бесконечности;
условиях (ss) при многократном введении лекарственного средства;
биодоступность) лекарственного средства, определяемая отношением
определяемая отношением AUC / AUC или AUC /
лекарственного средства превышает C ;
лекарственного средства превышает 75% от С ;
Оценка биоэквивалентности («фармакокинетической эквивалентности») лекарственных средств является основным видом медико-биологического контроля воспроизведенных (генерических) лекарственных средств, не отличающихся лекарственной формой и содержанием действующих веществ от соответствующих оригинальных лекарственных средств. Исследования биоэквивалентности позволяют сделать обоснованные заключения о качестве сравниваемых препаратов по относительно меньшему объему первичной информации и в более сжатые сроки, чем при проведении клинических исследований.
Настоящие Методические указания разработаны в соответствии со ст. 21 Конституции Российской Федерации, Федеральным законом «О лекарственных средствах» от 22.06.1998 N 86-ФЗ, Федеральным законом «О техническом регулировании» от 27.12.02 N 184-ФЗ, Правилами клинической практики в Российской Федерации (утверждены Приказом Минздрава России от 19.06.2003 N 266), Хельсинской декларацией Всемирной медицинской ассоциации (редакция, одобренная 52 сессией Генеральной ассамблеи в Эдинбурге, Шотландия, 2000).
В настоящей редакции Методических указаний уточнены формы лекарственных средств, для которых проводятся исследования биоэквивалентности, методики исследования биоэквивалентности и принципы анализа фармакокинетических данных при многократном введении лекарственных средств, процедура статистической оценки результатов исследования биоэквивалентности и т.д.
Биодоступность отражает количество неизмененного действующего вещества, достигающего системного кровотока (степень всасывания) относительно исходной дозы лекарственного средства.
Два лекарственных препарата являются биоэквивалентными, если они обеспечивают одинаковую биодоступность лекарственного средства.
2. Объекты исследований
Объектами исследований биоэквивалентности являются воспроизведенные лекарственные средства, предназначенные для приема внутрь, накожной аппликации, ректального введения, при условии, что их действие опосредовано появлением действующего вещества в системном кровотоке. Оценка биоэквивалентности проводится для всех лекарственных форм пролонгированного действия; форм, обеспечивающих немедленное высвобождение лекарственного средства при приеме внутрь (таблетки, капсулы, суспензии и др., за исключением растворов); трансдермальных терапевтических систем; ректальных и вагинальных суппозиториев, а также комбинированных лекарственных препаратов (по основным компонентам). Исследования биоэквивалентности не проводятся для лекарственных средств, предназначенных для введения путем ингаляции.
В качестве препарата сравнения следует использовать соответствующее оригинальное лекарственное средство, зарегистрированное в Российской Федерации.
Содержание действующего вещества в исследуемом лекарственном средстве и препарате сравнения не должно отличаться более чем на 5%.
Оценка биоэквивалентности всех лекарственных средств, за исключением психотропных и средств, применяемых при ВИЧ-инфекции, проводится на здоровых добровольцах. Соответствующие исследования психотропных средств, а также средств, применяемых при ВИЧ-инфекциях, могут выполняться на психических больных в период ремиссии заболевания и ВИЧ-инфицированных.
В случае, если после однократного приема лекарственного средства высока вероятность возникновения серьезных побочных эффектов, исследования по биоэквивалентности могут быть заменены оценкой относительной биодоступности препарата на крупных лабораторных животных (Приложение 5).
3.1. Критерии включения добровольцев в исследования
В качестве здоровых добровольцев могут привлекаться лица обоего пола в возрасте от 18 до 45 лет, отвечающие следующим критериям:
— верифицированный диагноз: «здоров» по данным стандартных клинических, лабораторных и инструментальных методов обследования;
— масса тела не выходит за пределы +/- 15% по весо-ростовому индексу Кетле;
3.2. Критерии исключения добровольцев из исследований:
— отягощенный аллергологический анамнез;
— хронические заболевания сердечно-сосудистой, бронхолегочной, нейроэндокринной системы, а также заболевания желудочно-кишечного тракта, печени, почек, крови;
— хирургические вмешательства на желудочно-кишечном тракте (за исключением аппендэктомии);
— острые инфекционные заболевания менее чем за 4 недели до начала исследования;
— регулярный прием лекарственных препаратов менее чем за 2 недели до начала исследования;
— прием лекарственных препаратов, оказывающих выраженное влияние на гемодинамику, функцию печени и др. (барбитураты, омепразол, циметидин и т.д.), менее чем за 30 дней до начала исследования;
— донорство (450 мл крови или плазмы и более) менее чем за 2 месяца до начала исследования;
— прием более чем 10 ед. алкоголя в неделю (1 ед. алкоголя эквивалентна 1/2 л пива, 200 мл вина или 50 мл спирта) или анамнестические сведения об алкоголизме, наркомании, злоупотреблении лекарственными препаратами;
— курение более 10 сигарет в день;
— участие в I фазе клинического испытания препаратов менее чем за 3 месяца до начала исследования.
3.3. Этические аспекты исследований биоэквивалентности
Участие здоровых испытуемых и больных в исследованиях биоэквивалентности лекарственных препаратов является добровольным. Доброволец (волонтер) имеет право отказаться от участия в проводимых исследованиях на любой его стадии. Этические нормы проведения испытаний биоэквивалентности регламентированы соответствующими документами. Этическую экспертизу клинических исследований биоэквивалентности лекарственных препаратов проводит Комитет по этике при федеральном органе контроля качества лекарственных средств. Добровольцы, включенные в исследование биоэквивалентности, подписывают письменное информированное согласие, один экземпляр которого выдается добровольцу наряду с «Информацией для добровольцев, участвующих в исследовании биоэквивалентности лекарственного препарата» (Приложения 1 и 2).
3.4. Группа исследователей-клиницистов
Для проведения исследований биоэквивалентности выделяются сотрудники, контролирующие состояние здоровья добровольцев, соблюдение режима, организацию питания, установку катетеров, отбор образцов крови и их обработку, оказывающие при необходимости экстренную медицинскую помощь. В состав группы обязательно должны входить врач-исследователь (1-2) и медицинская сестра (1-2).
3.5. Формирование банка добровольцев
Банк добровольцев формируется в соответствии с критериями включения в исследования и исключения из исследований, указанными в разделах 3.1. и 3.2.
В беседе с врачом-исследователем доброволец должен получить следующую информацию:
— наличие разрешения на проведение исследования;
— условия отбора проб крови;
— условия, в которых будет находиться доброволец во время исследования, пищевом и водном режиме;
— ограничения в приеме лекарств во время исследования;
— возможность оказания медицинской помощи во время и после исследования;
— условия страхования и вознаграждения.
Если доброволец включается в банк данных, на него заводится индивидуальная карта, где указываются:
— Ф.И.О., возраст, адрес, телефон, паспортные данные;
— медицинский анамнез (с указанием хронических заболеваний и аллергологический анамнез);
— перенесенные заболевания, по поводу которых доброволец находился на стационарном лечении.
В индивидуальной карте регистрируется участие добровольца во всех клинических исследованиях лекарственных средств.
3.6. Формирование группы добровольцев для проведения исследований биоэквивалентности конкретного лекарственного средства
За 1 неделю до начала испытаний добровольцы, привлекаемые к исследованиям конкретного лекарственного препарата, приглашаются в исследовательский центр. Врач-исследователь проводит с ними беседу, в ходе которой повторно собирается медицинский анамнез и проводится оценка соответствия добровольцев критериям включения в исследование (в соответствии с п.п. 3.1. и 3.2.).
Затем добровольцу предоставляется информация о:
— фармакологической группе, к которой относится исследуемое лекарственное средство;
— механизме его действия;
— показаниях к применению лекарственного средства;
— возможных нежелательных эффектах;
— пути введения и дозе;
— режиме питания перед началом исследования;
— режиме дня во время проведения исследования;
— времени прибытия в исследовательский центр;
— размере вознаграждения за участие в исследовании;
— условиях страхования, компенсации и лечения в случае причинения ущерба здоровью в связи с проведением исследования.
Добровольцу гарантируют, что при необходимости ему будет оказана квалифицированная медицинская помощь как во время, так и после проведения исследования биоэквивалентности, а также о том, что информация о нем, полученная в ходе исследований, будет иметь конфиденциальный характер. После этого доброволец для участия в исследовании должен подписать «Информированное согласие добровольца», копия которого выдается добровольцу наряду с «Информацией для добровольцев, участвующих в исследовании биоэквивалентности лекарственного препарата».
3.7. Скрининговое обследование добровольцев до начала исследований
После подписания информированного согласия проводится клиническое и параклиническое обследование добровольцев, включающее врачебный осмотр с учетом особенностей ожидаемого действия изучаемого лекарственного средства, а также следующие лабораторные тесты:
— клинический анализ крови;
— клинический анализ мочи;
— биохимический анализ крови (общий белок, билирубин, креатинин, аспартатаминотрансфераза, аланинаминотрансфераза, щелочная фосфатаза, глюкоза, холестерин);
— анализ крови на ВИЧ, сифилис, вирусный гепатит В и С;
Результаты обследования заносятся в индивидуальные карты добровольцев. По результатам клинического осмотра и лабораторных тестов врач-исследователь делает заключение, на основании которого доброволец допускается или не допускается к исследованию биоэквивалентности. Врач-исследователь составляет список добровольцев и передает его лицу, ответственному за проведение исследования. Проводится рандомизация добровольцев, после чего каждому из них присваивается номер, регистрируемый в индивидуальной карте.
3.8. Подготовка клинического блока
Клинический блок, где будут находиться добровольцы, должен включать следующие помещения: палаты для проживания добровольцев, процедурная, столовая, комната отдыха, душевая и туалет. Перед госпитализацией в указанных помещениях должна быть проведена санитарная обработка. Обязательным требованием к проведению исследований биоэквивалентности является наличие блока интенсивной терапии или реанимационного отделения.
3.9. Организация питания добровольцев
Добровольцы в ходе исследования должны получать доброкачественное и сбалансированное питание. Как правило, в меню включаются диетические блюда, исключается жирная и жареная пища и напитки, содержащие кофеин. Меню составляется накануне исследования и подается в пищеблок. Указывается время, к которому должна быть готова пища, название блюд и количество порций.
3.10. Отбор проб крови
При отборе проб крови должны соблюдаться следующие условия:
— кровь отбирается из локтевой вены через кубитальный катетер;
— первая порция крови (исходная, т.е. до приема препарата) берется утром, натощак, через 5-10 минут после установки катетера;
— испытуемый принимает исследуемый препарат или препарат сравнения, запивая его 200 мл кипяченой воды;
— время отбора последующих проб соответствует программе исследования;
— пробирки для отбора проб должны иметь маркировку с указанием шифра испытуемого, номера пробы и названия препарата;
— первый прием пищи должен быть не ранее чем через 4 ч после приема лекарственного средства;
— при возникновении экстремальной ситуации (ухудшение самочувствия, психические нарушения, желание испытуемого выйти из исследования) отбор проб прекращается;
— при возникновении непредвиденных ситуаций, исключающих возможность отбора крови в установленном временном интервале, работа с данным испытуемым продолжается, но шифрованная пробирка остается пустой;
— пробы крови с сопроводительным направлением, в котором указываются Ф.И.О. испытуемого, пол, возраст, масса тела, рост, соответствующие шифру на пробирке, предоставляются в фармакокинетическую лабораторию.
3.11. Динамическое наблюдение за добровольцами в ходе исследования
Динамическое наблюдение за добровольцами в период отбора образцов крови осуществляется врачом-исследователем и включает:
— клинический осмотр каждые 3-8 ч (в зависимости от фармакологических особенностей препарата);
— измерение уровня артериального давления и частоты сердечных сокращений.
Результаты обследования заносятся в индивидуальные карты добровольцев.
По окончании первого периода исследования после удаления катетеров проводится заключительный врачебный осмотр добровольцев. При отсутствии отклонений в состоянии здоровья добровольцев их отпускают домой до начала второго периода исследования.
Перед вторым периодом исследования проводится повторное обследование добровольцев, включающее врачебный осмотр, и, при необходимости, клинико-инструментальные исследования (ЭКГ и другие) и лабораторные тесты:
— клинический анализ крови;
— клинический анализ мочи;
— биохимический анализ крови;
На основании результатов повторного обследования врач-исследователь допускает или не допускает добровольцев ко второму периоду исследования.
Наблюдение за добровольцами в течение второго периода отбора образцов крови осуществляется так же, как и в первом периоде.
После завершения второго периода исследования биоэквивалентности проводится заключительный врачебный осмотр. При отсутствии отклонений в состоянии здоровья добровольцев их отпускают домой.
Для обеспечения безопасности исследования биоэквивалентности врачом-исследователем проводится мониторинг нежелательных явлений. Случаи возникновения нежелательных явлений регистрируются в индивидуальной карте добровольца и соответствующей форме.
3.12. Интервал между периодами исследований
В интервале времени между первым и вторым периодами исследований добровольцы должны соблюдать установленные правила. Продолжительность интервала определяется в зависимости от фармакокинетических свойств изучаемого препарата (обычно 7-14 дней).
3.13. Дублеры добровольцев
В ходе подготовки к исследованию осуществляется также подбор дублеров на случай замены выбывших из исследования добровольцев. Дублеры до начала исследования должны подписать информированное согласие и пройти обследование в том же объеме, что и добровольцы. Число дублеров составляет 4-6 человек.
4. Регламент фармакокинетического исследования
4.1. Общая схема исследований
4.2. Число испытуемых
В исследование должны быть включены испытуемые в количестве,
достаточном для обеспечения статистической значимости
исследования. При этом мощность статистического теста для проверки
биоэквивалентности должна поддерживаться на уровне не меньше 80%
для выявления 20%-ных различий между показателями сравнения.
Минимальное число включенных испытуемых 18 человек. Большее число
испытуемых может потребоваться для сравнения препаратов,
обладающих значительной вариабельностью фармакокинетических
параметров. При планировании эксперимента априорные значения
вариации основных показателей сравнения (AUC и С ), необходимые
для расчета числа испытуемых, могут быть оценены по результатам
сходных исследований, литературным данным, результатам пилотного
исследования, а предварительная оценка необходимого числа
испытуемых может быть получена с помощью таблицы 1.
ЧИСЛО ИСПЫТУЕМЫХ, НЕОБХОДИМЫХ ДЛЯ ОБЕСПЕЧЕНИЯ
80%-Й МОЩНОСТИ СТАТИСТИЧЕСКОГО КРИТЕРИЯ В СЛУЧАЕ
ЛОГ-НОРМАЛЬНОГО РАСПРЕДЕЛЕНИЯ (ГРАНИЦЫ ДОВЕРИТЕЛЬНОГО
ИНТЕРВАЛА 0,80-1,25; УРОВЕНЬ ЗНАЧИМОСТИ 5%)
│ Коэффициент вариации │ мю / мю │
│5 │12 │6 │4 │4 │4 │6 │8 │22 │
│7,5 │22 │8 │6 │6 │6 │8 │12 │44 │
│10,0 │36 │12 │8 │6 │8 │10 │20 │> 48│
│12,5 │> 48│16 │10 │8 │10 │14 │30 │> 48│
│15 │> 48│22 │12 │10 │12 │20 │42 │> 48│
│17,5 │> 48│30 │16 │14 │16 │26 │> 48│> 48│
│20,0 │> 48│38 │20 │16 │18 │32 │> 48│> 48│
│22,5 │> 48│46 │24 │20 │24 │40 │> 48│> 48│
│25,0 │> 48│> 48│28 │24 │28 │48 │> 48│> 48│
│27,5 │> 48│> 48│34 │28 │34 │> 48│> 48│> 48│
│30,0 │> 48│> 48│40 │32 │38 │> 48│> 48│> 48│
«ошибки» или остаточная внутрииндивидуальная вариация,
определяемая при дисперсионном анализе после логарифмической
трансформации значений показателя;
Если при проведении статистического сравнения уровень мощности теста оказался ниже 80%, в тех случаях, когда сравниваемые препараты оказались небиоэквивалентными, для принятия обоснованного заключения о небиоэквивалентности необходимо включить в исследование большее число испытуемых.
Подробнее эти вопросы рассмотрены в Приложении 3.
4.3. Доза лекарственного средства
Исследования биоэквивалентности проводятся с одной дозой воспроизведенного лекарственного средства в данной лекарственной форме, даже если для регистрации она заявлена в нескольких дозировках, при соблюдении следующих условий:
— качественный состав лекарственной формы, содержащей различное количество действующего вещества, одинаков;
— технология производства препаратов, содержащих различное количество лекарственного средства, одинакова;
— кинетика растворения лекарственного средства для препаратов с различной дозировкой должна быть эквивалентной (Приложение 4);
— фармакокинетика лекарственного средства линейна в терапевтическом диапазоне.
Биоэквивалентность препаратов в лекарственных формах пролонгированного действия оценивается для каждой дозы.
В исследованиях с многократным введением лекарственного препарата схема его дозирования не должна отличаться от рекомендованной(ых) в инструкции по медицинскому применению.
4.4. Интервал времени между приемом лекарственных средств
Интервал времени между приемом препаратов зависит от
длительности циркуляции лекарственного средства в организме,
определяемой периодом полувыведения (T ), и должен составлять не
4.5. Виды биологического материала, используемого для определения концентрации действующего вещества
При проведении исследований биоэквивалентности концентрация действующих веществ определяется в плазме, сыворотке или цельной крови.
4.6. Схема отбора проб
5. Аналитический метод
Для определения концентрации действующих веществ в плазме, сыворотке или цельной крови могут быть использованы различные методы (физико-химические, иммунологические, микробиологические и др.), обеспечивающие возможность получения достоверных лабораторных данных о концентрации действующего вещества при выбранных условиях фармакокинетического исследования, в частности его длительности, и отвечающие общим требованиям избирательности, точности, воспроизводимости.
Если вследствие пресистемной элиминации лекарственного средства оно не обнаруживается в крови в неизмененном состоянии и не обладает фармакологической активностью (пролекарство), необходимо определять концентрацию биологически активного метаболита.
6. Анализ фармакокинетических данных
6.1. Однократное введение лекарственных препаратов
времени ее достижения (t ) следует оценить внемодельными
испытуемого для каждого из изучаемых препаратов. Значения
параметров С и t оценивают как наибольшее из измеренных
значений концентрации и соответствующее время наблюдаемого
максимума. Величину AUC рассчитывают при помощи метода обычных
или логарифмических трапеций. Значения AUG определяют по
средства в последней пробе и константы элиминации соответственно.
Для вычисления C и k конечный (моноэкспоненциальный) участок
фармакокинетической кривой описывают с помощью нелинейного
При достаточной длительности наблюдения, когда AUC > 80%
AUC (см. раздел 4.6.), для оценки полноты всасывания исследуемого
6.2. Многократное введение лекарственных препаратов
В тех случаях, когда ввиду недостаточной чувствительности аналитического метода получить полноценные фармакокинетические профили после однократного введения лекарственного средства невозможно, а также когда внутрииндивидуальная вариабельность концентрации лекарственного средства при однократном введении выше, чем при его длительном введении, оценка биоэквивалентности препаратов проводится после их многократного введения.
В стационарных условиях (ss), реализующихся при повторяющемся
введении лекарственных препаратов в одинаковой дозе с одним и тем
же интервалом дозирования (тау), индивидуальные
фармакокинетические профили следует охарактеризовать значениями
дозирования после установления стационарного распределения
отнесенной к средней стационарной концентрации (C = AUC /
тау). Также вычисляют индивидуальные значения отношений AUC
и С для исследуемого препарата и препарата сравнения
(соответственно f’ и f»).
Для форм пролонгированного действия рассчитываются
продолжительность периода времени, в течение которого концентрация
лекарственного средства превышает среднее стационарное значение
7. Статистическая оценка биоэквивалентности
Оценка биоэквивалентности проводится по параметрам сравнения, выбранным в соответствии со схемой введения препарата (однократное и многократное введение) и его лекарственной формой (обычная или пролонгированного действия).
Статистический анализ проводят в предположении о
лог-нормальном распределении параметров AUC, C и C / AUC и
нормальном распределении остальных параметров, за исключением
средних значений параметров для исследуемого препарата и препарата
сравнения проводится на основе мультипликативной модели, а
доверительные интервалы строятся для отношений соответствующих
средних значений. После проведения логарифмического преобразования
эти показатели анализируются с помощью дисперсионного анализа
(ANOVA; параметрический метод).
Для обычной рандомизированной перекрестной схемы статистическая модель дисперсионного анализа должна включать следующие факторы, вносящие вклад в наблюдаемую вариацию данных:
— различия между препаратами;
— различия между испытуемыми (межиндивидуальные различия);
— последовательность приема препаратов;
Дисперсионный анализ применяется для проверки гипотез о статистической значимости вклада каждого из указанных факторов в наблюдаемую вариабельность. Полученная с помощью дисперсионного анализа оценка остаточной вариации используется при расчете доверительного интервала для отношения средних значений соответствующего параметра.
Процедура статистического сравнения состоит в вычислении
параметрических двусторонних 90%-ных доверительных интервалов для
отношений соответствующих средних значений для исследуемого
препарата и препарата сравнения. Препараты считаются
биоэквивалентными, если границы оцененного доверительного
вариабельностью, эти пределы составляют 75-133%.
8. Исключение резко выделяющихся наблюдений
Резко выделяющиеся наблюдения могут не приниматься в расчет при оценке биоэквивалентности при условии, что справедливость исключения этих данных доказана.
9. Группа исследователей-специалистов в области фармакокинетики
Для проведения исследований выделяются сотрудники, обладающие профессиональным опытом в области количественного анализа лекарственных веществ в биоматериале и математического анализа фармакокинетических данных.
10. Протокол проведения исследований
В протоколе должны быть отражены:
— сведения об изучаемых лекарственных средствах (лекарственная форма, содержание действующего вещества, фирма-производитель);
— сведения об испытуемых и об их количестве;
— доза и режим дозирования;
— интервал времени между приемом препаратов;
— биоматериал, в котором предполагается определять концентрацию действующего вещества;
— схема отбора проб, условия их хранения;
— сведения об аналитическом методе;
— сведения о методах фармакокинетического анализа;
— сведения о критериях биоэквивалентности.
11. Отчетная документация
В отчете об исследованиях биоэквивалентности должны быть представлены:
— утвержденный протокол проведения исследований;
— названия исследуемых лекарственных средств;
— номера серий исследуемых препаратов, данные о сроке их годности;
— сведения о содержании действующего вещества в изученных препаратах;
— демографические и антропометрические данные испытуемых;
— клинические данные (если исследование биоэквивалентности проведено на больных), в которых указывается анамнез заболевания, диагноз, характер поражения внутренних органов и их функциональное состояние на момент проведения исследований;
— способ введения лекарственного препарата и дозы;
— методика отбора биоматериала и его предварительной обработки, условия хранения проб;
— описание аналитического метода, включающее метрологические характеристики и демонстрационные хроматограммы, если использованы хроматографические методы;
— описание процедур фармакокинетического анализа и оценки биоэквивалентности с указанием использованных программных средств;
— результаты определения содержания действующего вещества в биопробах, соответствующие средние значения и величины стандартных отклонений;
— индивидуальные фармакокинетические профили;
— усредненные фармакокинетические профили;
— индивидуальные значения параметров фармакокинетики, соответствующие средние значения и величины стандартных отклонений;
— средние геометрические значения лог-нормально распределенных параметров фармакокинетики, соответствующие интервальные оценки;
— результаты дисперсионного анализа, значения соответствующих коэффициентов вариации;
— статистические критерии, использованные для оценки биоэквивалентности, и результаты этой оценки;
1. Соловьев В.Н., Фирсов А.А., Филов В.А. Фармакокинетика (руководство). М.: Медицина, 1980. 423 с.
3. Bioequivalence Assessment. Methods and Applications. Ed. Steinijans V.W. Int. J. Clin. Pharmacol. Ther. and Toxicol., Vol. 30 (Suppl. 1), 1992, pp. 1-66.
4. Investigation of Bioavailability and Bioequivalence. Commission of the European Communities, III/54/89-EN, December 1991, pp. 1-20.
5. In vivo bioequivalence guidances. U.S. Pharmacopeia 24-NF 19, Supplement 2, 2000, 1090, pp. 2056-2098.
6. Note for Guidance on the Investigation of Bioavailability and Bioequivalence. The European Agency for the Evaluation of Medical Products, Committee for Proprietary Medicinal Products, London, July 2001, pp. 1-18.
7. Statistical Approaches to Establishing Bioequivalence. U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research, January 2001.
8. Waiver of in vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research, August 2000.
9. Diletti E., Hauschke D., Steinijans V.W. Int. J. Clin. Pharmacol. Ther. and Toxicol. Sample size determination for bioequivalence assessment by means of confidence intervals. 1991, Vol. 29, pp. 1-8.
10. Hauschke D., Steinijans V.W., Diletti E.A. Distribution-free Procedure for the Statistical Analysis of Bioequivalence Studies. Int. J. Clin. Pharmacol. Ther. Toxicol. 1990, Vol. 28, pp. 72-78.
11. Hills M., Armitage P. The Two-Period Cross-Over Clinical Trial. Br. J. Clin. Pharmac. 1979, Vol. 8, pp. 7-20.
12. Lund R.E. Tables for an Approximate Test for Outliers in Linear Models. Technometrics. 1975, Vol. 17, pp. 473-476.
13. Schuirmann D.J. A comparison of the two one-side tests procedure and the power approach for assessing the equivalence of average bioavailability. J. Pharmacokinet. Biopharm. 1987, Vol. 15, pp. 657-680.
14. Shein-Shung Chow, Jen-Pei Liu. Design and Analysis of Bioavailability and Bioequivalent Studies. Marcel Dekker, New York, 1992.
15. Steinijans V.W., Diletti E. Statistical Analysis of Bioavailability Studies: Parametric and Nonparametric Confidence Intervals. Eur. J. Clin. Pharmacol. 1983, Vol. 24, pp. 127-136.
16. Steinijans V.W., Sauter R., Jonkman H.G. et al. Bioequivalence Studies: Single vs Multiple Dose. Int. J. Clin. Pharmacol. Ther. Toxicol. 1989, Vol. 27, pp. 261-266.
17. Wijnand H.P. Bioequivalence assessment of drug formulations. Non-parametric versus parametric analysis. Doctor’s thesis. Leiden, 1994, pp. 1-60.
СОСТАВЛЕНИЯ ИНФОРМАЦИИ ДЛЯ ДОБРОВОЛЬЦА
С ФОРМОЙ ИНФОРМИРОВАННОГО СОГЛАСИЯ
Форма информированного согласия добровольца (волонтера)
на участие в исследованиях биоэквивалентности
Я, ___________, проживающий по адресу ________________, тел. ________, паспорт: серия _______ N __________, выдан ______________, ознакомился с информацией о целях предстоящего исследования биоэквивалентности препарата _____________________ и получил копию данного документа. Я имел возможность обсудить с врачом-исследователем все интересующие меня вопросы и получил удовлетворяющие меня ответы.
Я предупрежден о том, что исследование биоэквивалентности лекарственного препарата ____________ будет вызывать некоторый дискомфорт, а также не исключена возможность вредного влияния изучаемого лекарственного препарата на мое здоровье или самочувствие.
Я добровольно соглашаюсь принять участие в исследовании биоэквивалентности вышеупомянутого лекарственного препарата, извещен, что имею право отказаться или в любой момент прекратить участие в данном исследовании, что не повлечет за собой изменения отношения ко мне медицинского персонала.
В случае моего решения о прекращении моего участия в исследовании биоэквивалентности я обязуюсь информировать об этом врача-исследователя для того, чтобы предоставить ему возможность оценить мое состояние и дать необходимые рекомендации.
Я согласен выполнять инструкции, добросовестно сотрудничать с врачом-исследователем и немедленно сообщать ему о любого рода изменениях моего здоровья.
Я извещен, что информация, полученная в ходе исследования, является конфиденциальной. Я согласен с тем, чтобы она использовалась в полной мере и передавалась в регуляторные органы и официальные медицинские инстанции, а также уполномоченным лицам и представителям спонсора.
Я извещен, что, если моему здоровью будет причинен ущерб, связанный с моим участием в исследовании, страховая компания выплатит мне компенсацию. Сумма компенсации может быть пересмотрена в случае моей вины в возникновении ухудшения здоровья.
Ф.И.О. добровольца _____________________________
Подпись добровольца: ___________________ Дата _________________
Ф.И.О. врача-исследователя _____________________
Подпись врача-исследователя: ___________ Дата _________________
ОТЧЕТ О СЕРЬЕЗНОМ НЕЖЕЛАТЕЛЬНОМ ЯВЛЕНИИ
Информация для добровольцев, участвующих в исследованиях биоэквивалентности лекарственного средства
Исследования проводятся на здоровых добровольцах.
Для определения состояния здоровья добровольца проводится полное обследование, включающее клинические и лабораторные тесты (анализы крови, мочи, исследование на ВИЧ, RW, вирусный гепатит В и С, ЭКГ). Если нет противопоказаний, доброволец включается в группу испытуемых.
Доброволец подписывает информированное письменное согласие, в котором он дает свое согласие на участие в исследовании.
Доброволец обязуется выполнять следующие правила:
— в течение недели до исследования и во время исследования доброволец не принимает лекарственные препараты. При необходимости приема лекарственных средств следует проконсультироваться с врачом-исследователем;
— накануне исследования и во время исследования доброволец не употребляет кофеинсодержащие продукты (чай, кофе и др.);
— за семь дней до исследования и во время проведения исследования доброволец не употребляет алкоголь и ограничивает себя в курении;
— накануне исследования последний прием пищи должен заканчиваться не позднее 19 часов, ужин не должен включать жирную пищу;
— исследование проводится в стационарных условиях, доброволец обязан соблюдать правила пребывания в стационаре.
Принципиальная схема исследования биоэквивалентности лекарственного средства:
— во время исследования добровольцы находятся в специальном блоке, где им созданы необходимые гигиенические условия и будет предложено 3-4-разовое питание. В случае необходимости добровольцам оказывается квалифицированная медицинская помощь;
— исследование биоэквивалентности состоит из двух равнозначных периодов.
Каждый из этих периодов начинается в ____ часов утра, когда добровольцу устанавливают в одну из вен руки катетер, через который в процессе исследования по заранее представленной схеме будет проводиться отбор крови. Всего будет взято не более 150-200 мл крови. При необходимости (в случае тромбоза катетера, выхода катетера из вены и др.) катетер будет переставлен в другую вену;
— после первого отбора крови доброволец должен принять исследуемый лекарственный препарат или его аналог. Последовательность приема препаратов будет случайной;
— после отбора последней пробы крови у добровольца будет удален внутривенный катетер и он будет отпущен домой до следующего периода исследования;
— в случае возникновения нежелательных явлений добровольцу будет оказана квалифицированная медицинская помощь;
— перерыв между периодами исследований составляет ___ дней. Доброволец в промежутке между ними должен соблюдать вышеизложенные требования;
— в случае неявки добровольца на 2-й период исследований он исключается из исследований и лишается права на вознаграждение.
Условиями прекращения участия в исследовании добровольца являются:
— несоблюдение правил проведения исследования;
На время исследования добровольцы страхуются на случай возникновения возможных нежелательных явлений, связанных с применением лекарственных препаратов.
При возникновении любых вопросов, связанных с проведением исследования или изменением в состоянии здоровья, следует обращаться к врачу _____________
Контактный телефон ____________________________
ВОПРОСЫ ПЛАНИРОВАНИЯ ИССЛЕДОВАНИЙ
БИОЭКВИВАЛЕНТНОСТИ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
для испытуемого препарата и препарата сравнения соответственно.
Гипотеза о биоэквивалентности может быть сформулирована в терминах
отношения сравниваемых параметров или в терминах их абсолютных
различий. Для параметров, связанных с определением концентрации
сравнение отношений (в предположении об лог-нормальном
нижняя и верхняя принятые допустимые границы биоэквивалентности,
нулевая гипотеза об отсутствии эквивалентности может быть записана
а соответствующая альтернативная гипотеза о наличии
биоэквивалентности формулируется как:
H : Q В случае проведения логарифмического преобразования формулировка гипотез также трансформируется:
H’ : дельта Вывод об эквивалентности в результате тестирования гипотез может быть сделан, если 100 x (1-2 x альфа)% доверительный интервал для разницы (или отношения) средних значений полностью располагается внутри выбранного интервала эквивалентности.
Необходимость определения требуемого числа испытуемых диктуется экономическими и этическими соображениями. Число испытуемых, необходимое для получения статистически значимого заключения о биоэквивалентности исследуемых препаратов, зависит от уровня значимости (альфа), мощности теста (1-бета), допустимой величины различий эпсилон и величины дисперсии данных сигма. Три первые величины регламентированы настоящими рекомендациями. Вариация данных оценивается либо по прошлому опыту проведения аналогичных исследований, либо по литературным данным, либо по пилотному исследованию. Чем более вариабельны данные, тем больше испытуемых потребуется для демонстрации биоэквивалентности лекарственных препаратов.
При проведении исследований биоэквивалентности в соответствии
с обычным перекрестным дизайном в предположении об отсутствии
влияния периода наблюдения вычисление доверительных интервалов
базируется на парном критерии Стьюдента. Поэтому самым простым
способом оценки необходимого числа испытуемых (n) будет расчет по
следующей формуле в предположении равенства средних значений мю =
2сигма (t(f, альфа) + t(f, бета))
значениями, число степеней свободы f = n-1 в случае вычисления
доверительных интервалов на базе парного критерия Стьюдента, f =
n-2 в случае, если учитывается влияние периода наблюдений и
доверительный интервал строится на базе подходящей процедуры
дисперсного анализа. В любом случае получается, что для
определения по таблицам значения коэффициента Стьюдента t(f,
альфа) необходимо знать число включенных пациентов n, а именно его
мы и пытаемся рассчитать по формуле. Для разрешения этого
противоречия обычно применяют итерационную процедуру, а в качестве
первоначальной оценки значения коэффициента Стьюдента берется его
соответствующее значение при бесконечном числе степеней свободы,
то есть соответствующее значение стандартного нормального
влияния периода наблюдений обычно используют среднее квадратичное
отклонение, оцененное с помощью анализа вариации для перекрестного
квадрата «ошибки» или остаточной внутрииндивидуальной вариации).
Последнее уравнение может быть модифицировано и выражено в терминах коэффициента вариации CV = сигма / мю и ОМЕГА = эпсилон / мю как:
CV x (t(f, альфа) + t(f, бета))
В соответствии с настоящими рекомендациями при этом определении величина ОМЕГА не должна превышать значение 0,2 (20%).
Приведенные формулы для оценки необходимого числа испытуемых
применимы как в случае аддитивной, так и в случае
мультипликативной модели данных. Различия заключаются в оценке
величины дисперсии сигма или коэффициента вариации CV. Так, в
случае предположения о лог-нормальном распределении данных и
сравнения отношения средних значений коэффициент вариации
полученному в результате дисперсионного анализа для
CV достаточно мал, CV
Рассмотрим случай, когда известно, что предположение о
равенстве средних значений показателей мю = мю = мю нарушается.
процедуры проверки двух односторонних тестов симметричны
относительно нуля, рассмотрим случай 0 Тогда необходимое число испытуемых в исследовании для поддержания мощности на уровне 80% и допустимых различиях ОМЕГА% (обычно 20%) определяется по формуле:
CV x (t(f, альфа) + t(f, бета))
формулы значения n опять приводит к необходимости применения
итерационной процедуры поиска.
В случае нарушения предположения о равенстве средних значений последняя формула может быть записана как:
Мощность статистического теста должна быть не меньше 80%.
ИССЛЕДОВАНИЯ КИНЕТИКИ РАСТВОРЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Для лекарственного средства, заявленного для регистрации в нескольких дозировках, проводится оценка эквивалентности кинетики перехода действующего вещества в раствор для каждой дозировки. Методически степень перехода действующего вещества в раствор определяют в условиях, описанных для данного препарата в соответствующей фармакопейной статье, для нескольких (не менее трех) временных точек, расположенных равномерно в интервале времени исследования. Последняя точка профиля должна соответствовать моменту перехода в раствор не менее 90% лекарственного средства или фазе насыщения процесса.
Эквивалентность кинетики растворения лекарственного средства
оценивают исходя из фактора сходимости (f ), который рассчитывают
из препарата сравнения в i-той временной точке (в среднем, в
из исследуемого препарата в i-той временной точке (в среднем, в
Оценку эквивалентности кинетики растворения проводят при следующих условиях:
— для каждой временной точки для каждого препарата проводят не менее 12 параллельных определений;
— только одно из рассчитанных средних значений для каждого препарата может быть больше 85%;
— величина стандартного отклонения для каждого среднего значения, за исключением первой временной точки, не должна быть больше 10%.
Кинетика растворения лекарственного средства считается
эквивалентной, если значение f лежит в пределах от 50 до 100. В
том случае, когда более 85% лекарственного средства переходит в
раствор в течение 15 мин., кинетика растворения считается
эквивалентной без математической оценки.
ЛЕКАРСТВЕННЫХ СРЕДСТВ, ДЛЯ КОТОРЫХ ДОПУСКАЕТСЯ
ПРОВЕДЕНИЕ ИССЛЕДОВАНИЙ БИОЭКВИВАЛЕНТНОСТИ
НА КРУПНЫХ ЛАБОРАТОРНЫХ ЖИВОТНЫХ
│ │терапев- │непатентованное│min, │max, │достижения│ 1/2 │
│ │тическая │ наименование │ мг │ мг │макси- │ │
│1 │Нейролеп- │Хлорпромазин │25 │75 │ │ │
│ │ │Левомепромазин │25 │50 │ 1-3 │ 15-78 │
│ │ │Галоперидол │1,5 │4,5 │ │ 13-40 │
│ │ │Сульпирид │200 │600 │ 4,5 │ 7 │
│2 │Противо- │Этосуксимид │250 │ │ │ │
│ │препараты │Карбамазепин │100 │200 │ 4-8 │ 15 │
│ │ │Вальпроевая │150 │ │ 2-3 │ 8-20 │
│3 │Средства, │Фенобарбитал │100 │ │ │ │
│ │при │Нитразепам │5 │10 │ 1-4 │ 26 │
│ │сна │Зопиклон │7,5 │15 │ │ 3,5-6 │
│ │ │Мидазолам │7,5 │15 │ 30 мин. │ 1,5-2,5 │
│4 │Миорелак- │Толперизон │50 │100 │ │ │
│ │ │Баклофен │10 │25 │ 2-3 │ 4 │
│5 │Антидепрес-│Амитриптилин │10 │25 │ │ 9-25 │



